Scleroderma Unit, Referral Center for Systemic Autoimmune Diseases, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico di Milano, Milan, Italy
GENYO, Centre for Genomics and Oncological Research Pfizer, University of Granada, Andalusian Regional Government, PTS GRANADA, Granada, Spain.
Ann Rheum Dis. 2020 Sep;79(9):1218-1226. doi: 10.1136/annrheumdis-2020-217116. Epub 2020 Jun 19.
The analysis of annotated transcripts from genome-wide expression studies may help to understand the pathogenesis of complex diseases, such as systemic sclerosis (SSc). We performed a whole blood (WB) transcriptome analysis on RNA collected in the context of the European PRECISESADS project, aiming at characterising the pathways that differentiate SSc from controls and that are reproducible in geographically diverse populations.
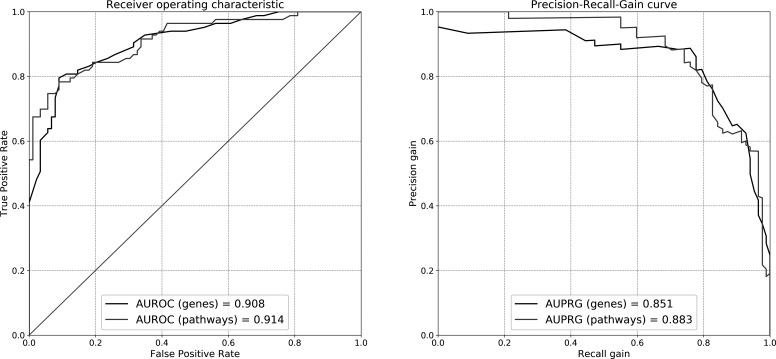
Samples from 162 patients and 252 controls were collected in RNA stabilisers. Cases and controls were divided into a discovery (n=79+163; Southern Europe) and validation cohort (n=83+89; Central-Western Europe). RNA sequencing was performed by an Illumina assay. Functional annotations of Reactome pathways were performed with the Functional Analysis of Individual Microarray Expression (FAIME) algorithm. In parallel, immunophenotyping of 28 circulating cell populations was performed. We tested the presence of differentially expressed genes/pathways and the correlation between absolute cell counts and RNA transcripts/FAIME scores in regression models. Results significant in both populations were considered as replicated.
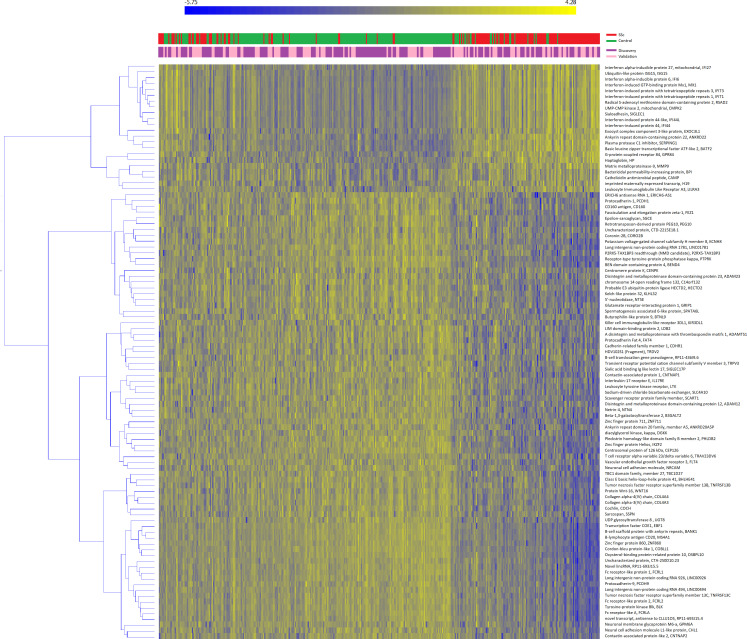
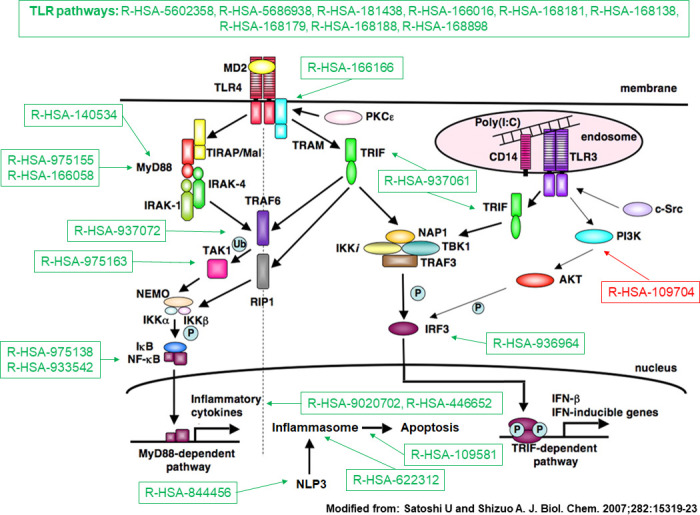
Overall, 15 224 genes and 1277 functional pathways were available; of these, 99 and 225 were significant in both sets. Among replicated pathways, we found a deregulation in type-I interferon, Toll-like receptor cascade, tumour suppressor p53 protein function, platelet degranulation and activation. RNA transcripts or FAIME scores were jointly correlated with cell subtypes with strong geographical differences; neutrophils were the major determinant of gene expression in SSc-WB samples.
We discovered a set of differentially expressed genes/pathways validated in two independent sets of patients with SSc, highlighting a number of deregulated processes that have relevance for the pathogenesis of autoimmunity and SSc.
对全基因组表达研究的注释转录本进行分析,有助于理解系统性硬化症(SSc)等复杂疾病的发病机制。我们在欧洲 PRECISESADS 项目背景下对全血(WB)转录组进行了分析,旨在确定区分 SSc 与对照的途径,这些途径在地理上多样化的人群中具有可重复性。
在 RNA 稳定剂中收集了 162 名患者和 252 名对照者的样本。病例和对照者被分为发现队列(n=79+163;南欧)和验证队列(n=83+89;中欧和西欧)。通过 Illumina 分析进行 RNA 测序。使用功能分析个体微阵列表达(FAIME)算法对 Reactome 途径的功能注释进行分析。同时,对 28 种循环细胞群进行免疫表型分析。我们在回归模型中测试了差异表达基因/途径的存在以及绝对细胞计数与 RNA 转录物/FAIME 评分之间的相关性。在两个群体中均显著的结果被认为是可复制的。
总体而言,有 15224 个基因和 1277 个功能途径可用;其中,99 个和 225 个在两组中均具有显著性。在复制的途径中,我们发现 I 型干扰素、Toll 样受体级联、肿瘤抑制因子 p53 蛋白功能、血小板脱颗粒和激活等途径出现失调。RNA 转录物或 FAIME 评分与具有强烈地理差异的细胞亚型共同相关;中性粒细胞是 SSc-WB 样本基因表达的主要决定因素。
我们发现了一组在两个独立的 SSc 患者队列中得到验证的差异表达基因/途径,突出了许多与自身免疫和 SSc 发病机制相关的失调过程。