Department of Computer Science, University of California, Irvine, CA, 92617, USA.
Computational Biology and Bioinformatics Program, Yale University, New Haven, CT, 06520, USA.
BMC Bioinformatics. 2020 Jul 2;21(1):281. doi: 10.1186/s12859-020-03605-3.
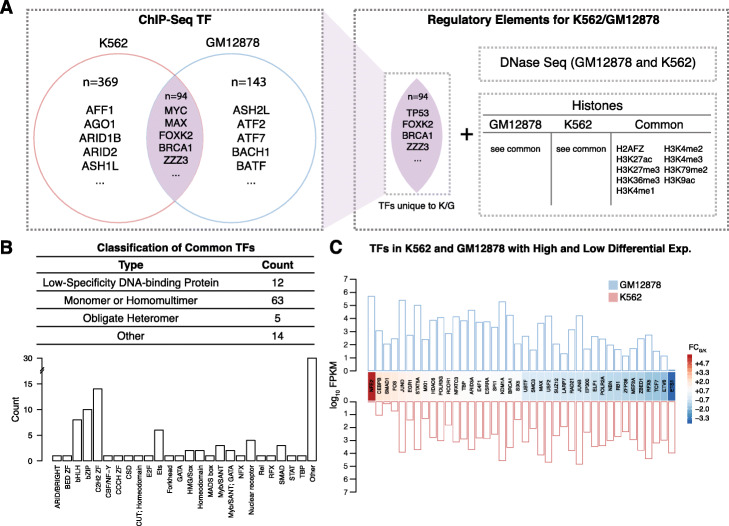
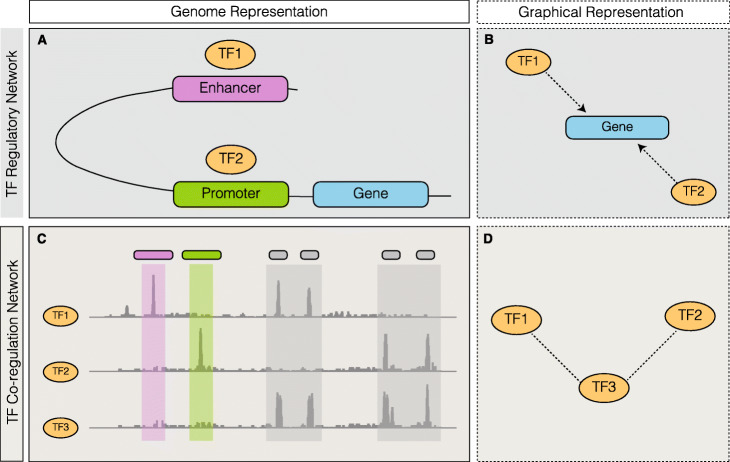
During transcription, numerous transcription factors (TFs) bind to targets in a highly coordinated manner to control the gene expression. Alterations in groups of TF-binding profiles (i.e. "co-binding changes") can affect the co-regulating associations between TFs (i.e. "rewiring the co-regulator network"). This, in turn, can potentially drive downstream expression changes, phenotypic variation, and even disease. However, quantification of co-regulatory network rewiring has not been comprehensively studied.
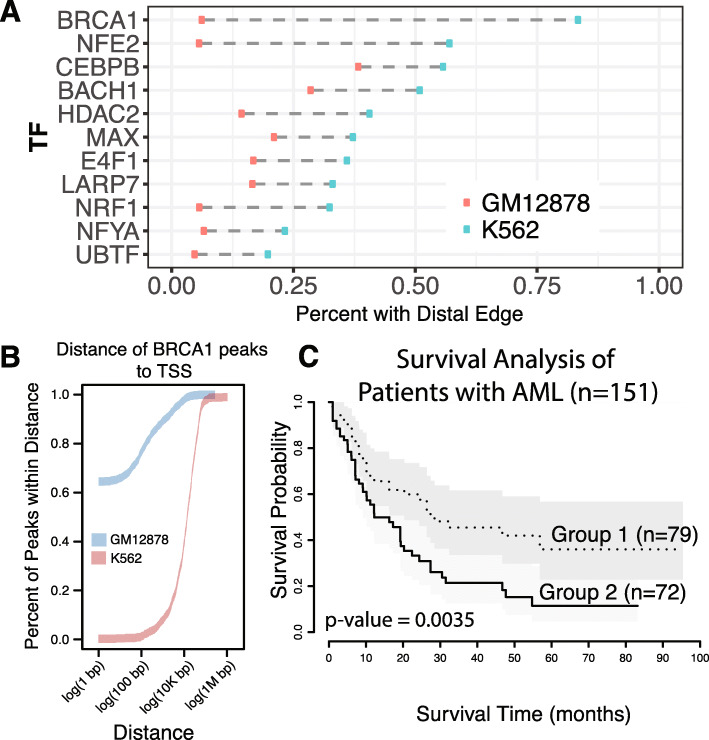
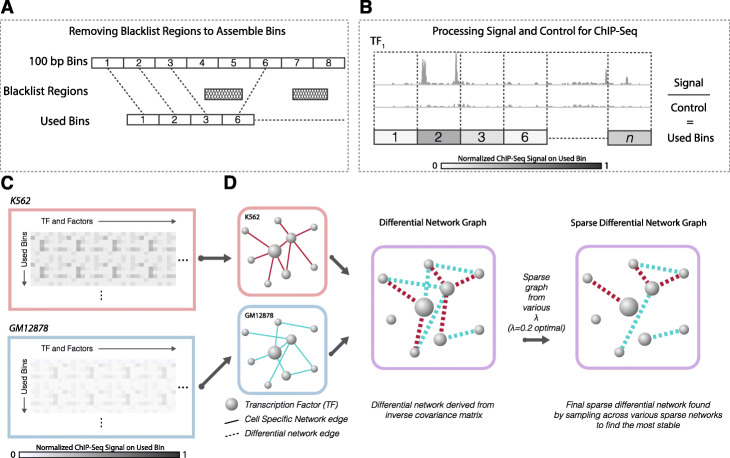
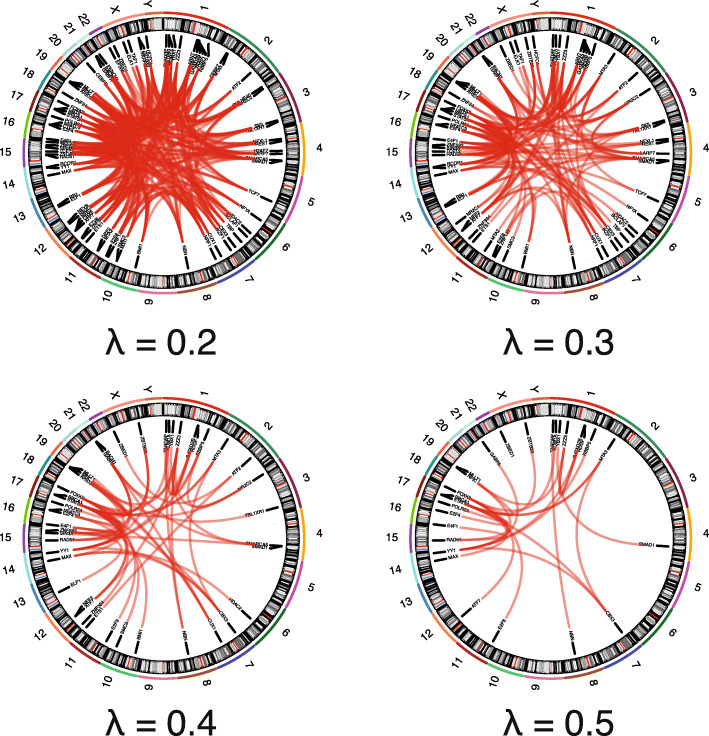
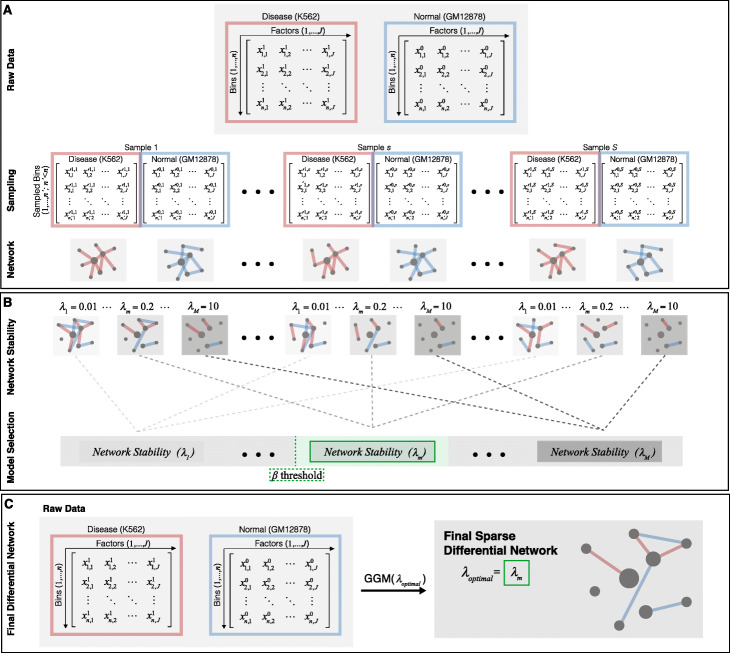
To address this, we propose DiNeR, a computational method to directly construct a differential TF co-regulation network from paired disease-to-normal ChIP-seq data. Specifically, DiNeR uses a graphical model to capture the gained and lost edges in the co-regulation network. Then, it adopts a stability-based, sparsity-tuning criterion -- by sub-sampling the complete binding profiles to remove spurious edges -- to report only significant co-regulation alterations. Finally, DiNeR highlights hubs in the resultant differential network as key TFs associated with disease. We assembled genome-wide binding profiles of 104 TFs in the K562 and GM12878 cell lines, which loosely model the transition between normal and cancerous states in chronic myeloid leukemia (CML). In total, we identified 351 significantly altered TF co-regulation pairs. In particular, we found that the co-binding of the tumor suppressor BRCA1 and RNA polymerase II, a well-known transcriptional pair in healthy cells, was disrupted in tumors. Thus, DiNeR successfully extracted hub regulators and discovered well-known risk genes.
Our method DiNeR makes it possible to quantify changes in co-regulatory networks and identify alterations to TF co-binding patterns, highlighting key disease regulators. Our method DiNeR makes it possible to quantify changes in co-regulatory networks and identify alterations to TF co-binding patterns, highlighting key disease regulators.
在转录过程中,许多转录因子(TF)以高度协调的方式结合到靶标上,以控制基因表达。TF 结合谱(即“共结合变化”)组的改变会影响 TF 之间的共调节关联(即“重新布线共调节网络”)。反过来,这可能会导致下游表达变化、表型变异,甚至疾病。然而,共调节网络重新布线的定量尚未得到全面研究。
为了解决这个问题,我们提出了 DiNeR,这是一种从配对的疾病到正常 ChIP-seq 数据中直接构建差异 TF 共调控网络的计算方法。具体来说,DiNeR 使用图形模型来捕获共调控网络中获得和丢失的边。然后,它采用基于稳定性、稀疏性调整的标准——通过对完整的结合谱进行子采样以去除虚假边缘——仅报告显著的共调控变化。最后,DiNeR 突出了差异网络中作为与疾病相关的关键 TF 的枢纽。我们组装了 K562 和 GM12878 细胞系中 104 个 TF 的全基因组结合谱,这些细胞系松散地模拟了慢性髓细胞白血病(CML)中正常和癌变状态之间的转变。总的来说,我们确定了 351 个显著改变的 TF 共调控对。特别是,我们发现肿瘤抑制因子 BRCA1 和 RNA 聚合酶 II(健康细胞中众所周知的转录对)的共结合在肿瘤中被破坏。因此,DiNeR 成功提取了枢纽调节剂并发现了众所周知的风险基因。
我们的方法 DiNeR 使得量化共调控网络的变化和识别 TF 共结合模式的改变成为可能,突出了关键的疾病调节剂。