Perrigue Patrick M, Rakoczy Magdalena, Pawlicka Kamila P, Belter Agnieszka, Giel-Pietraszuk Małgorzata, Naskręt-Barciszewska Mirosława, Barciszewski Jan, Figlerowicz Marek
Institute of Bioorganic Chemistry of the Polish Academy of Sciences, Zygmunta Noskowskiego 12/14, 61-704 Poznań, Poland.
NanoBioMed Center, Adam Mickiewicz University, Wszechnicy Piastowskiej 3, 61-614 Poznań, Poland.
Cancers (Basel). 2020 Jun 30;12(7):1745. doi: 10.3390/cancers12071745.
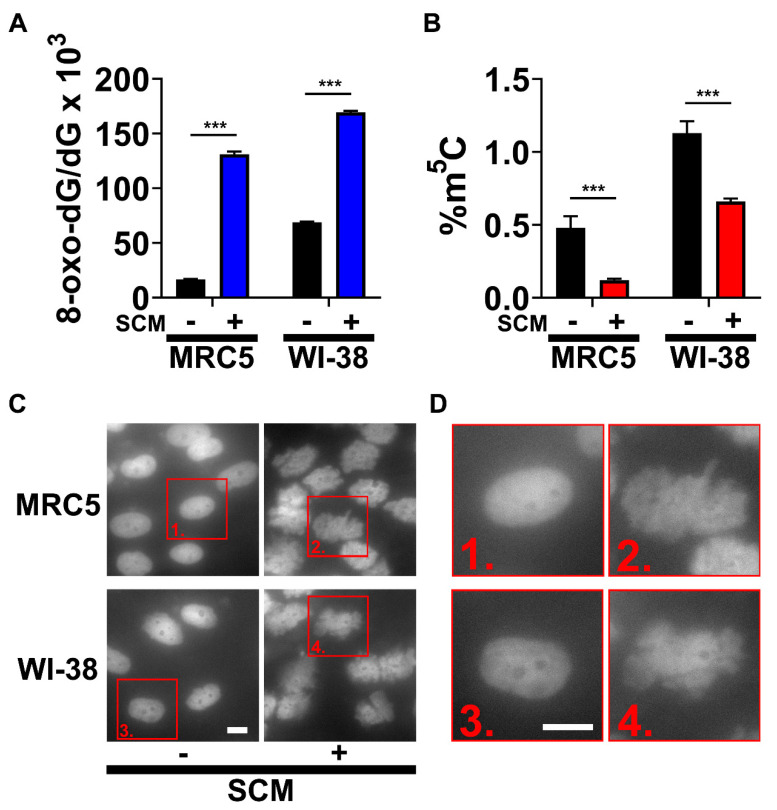
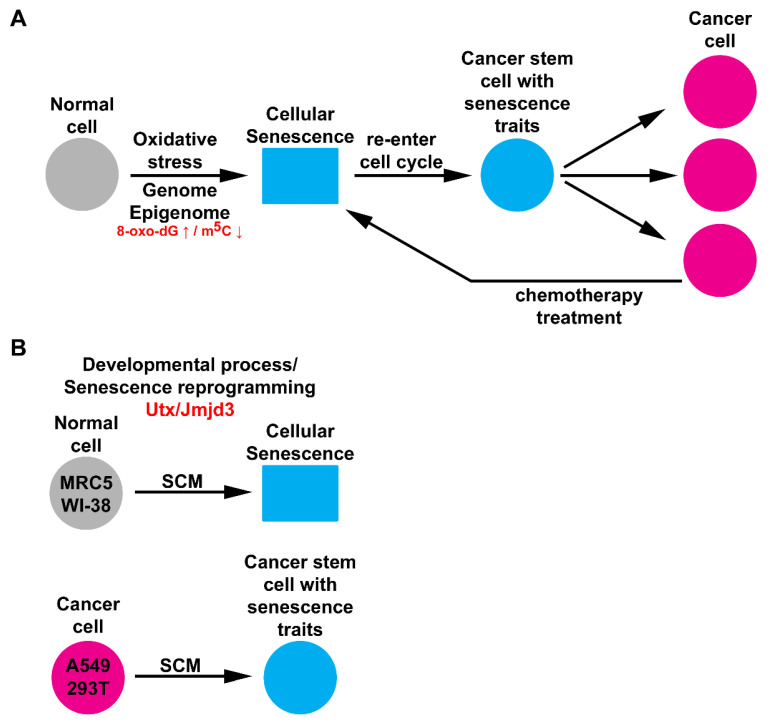
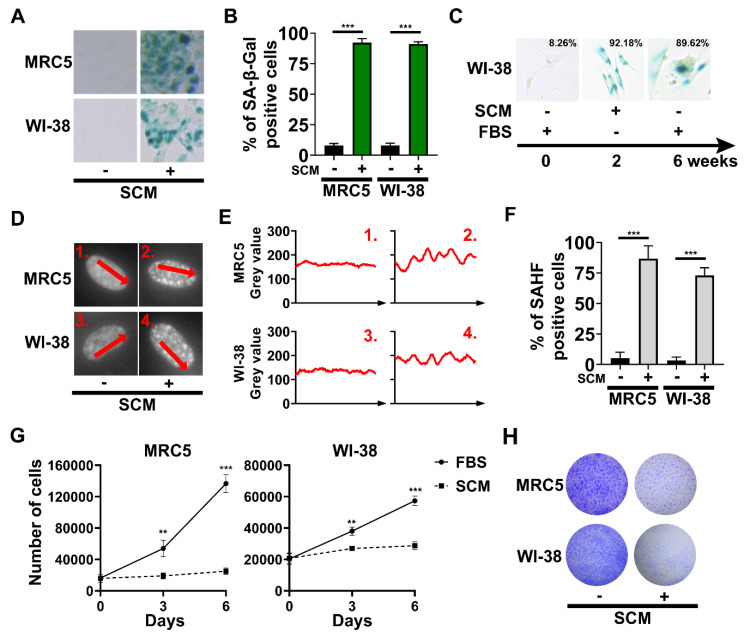
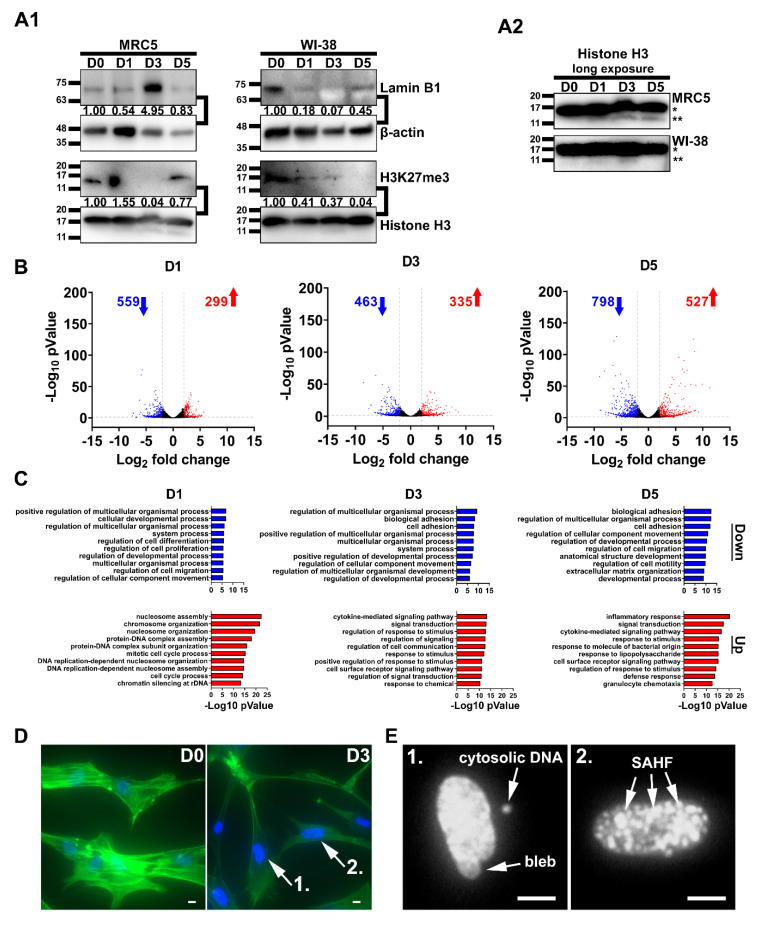
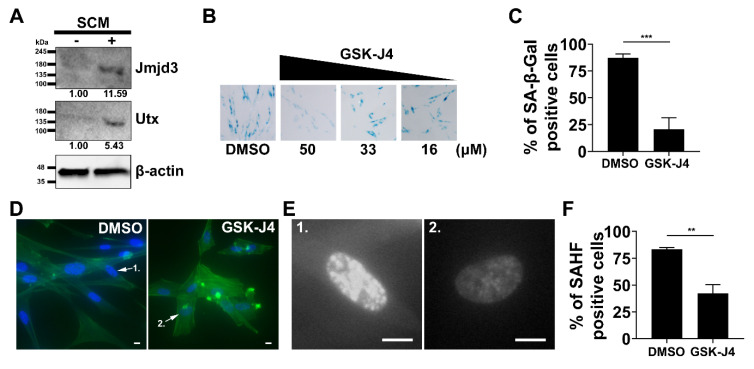
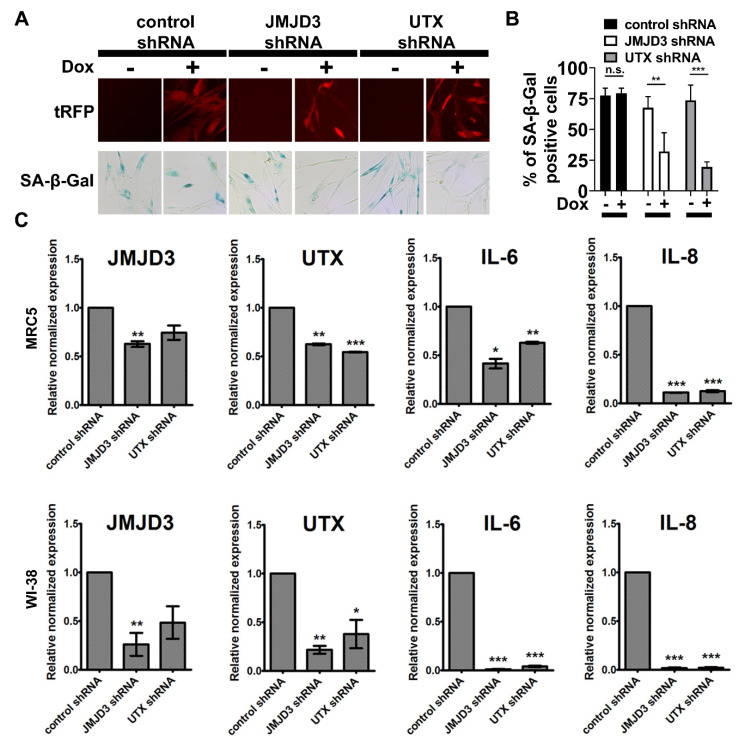
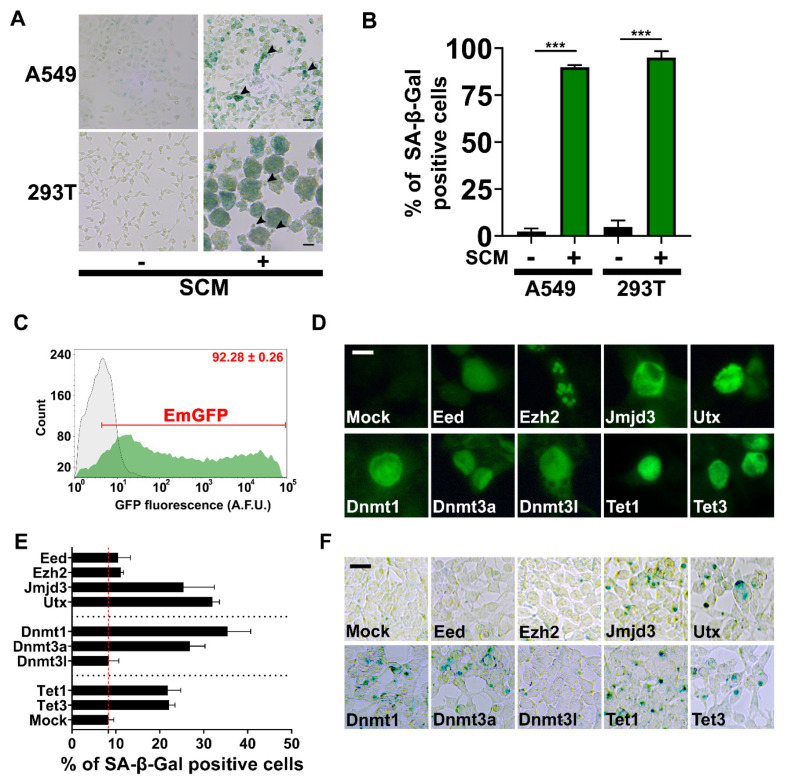
Cellular senescence is a tumor-suppressive mechanism blocking cell proliferation in response to stress. However, recent evidence suggests that senescent tumor cells can re-enter the cell cycle to become cancer stem cells, leading to relapse after cancer chemotherapy treatment. Understanding how the senescence reprogramming process is a precursor to cancer stem cell formation is of great medical importance. To study the interplay between senescence, stemness, and cancer, we applied a stem cell medium (SCM) to human embryonic fibroblasts (MRC5 and WI-38) and cancer cell lines (A549 and 293T). MRC5 and WI-38 cells treated with SCM showed symptoms of oxidative stress and became senescent. Transcriptome analysis over a time course of SCM-induced senescence, revealed a developmental process overlapping with the upregulation of genes for growth arrest and the senescence-associated secretory phenotype (SASP). We demonstrate that histone demethylases jumonji domain-containing protein D3 (Jmjd3) and ubiquitously transcribed tetratricopeptide repeat, X chromosome (Utx), which operate by remodeling chromatin structure, are implicated in the senescence reprogramming process to block stem cell formation in fibroblasts. In contrast, A549 and 293T cells cultured in SCM were converted to cancer stem cells that displayed the phenotype of senescence uncoupled from growth arrest. The direct overexpression of DNA methyltransferases (Dnmt1 and Dnmt3A), ten-eleven translocation methylcytosine dioxygenases (Tet1 and Tet3), Jmjd3, and Utx proteins could activate senescence-associated beta-galactosidase (SA-β-gal) activity in 293T cells, suggesting that epigenetic alteration and chromatin remodeling factors trigger the senescence response. Overall, our study suggests that chromatin machinery controlling senescence reprogramming is significant in cancer stem cell formation.
细胞衰老作为一种肿瘤抑制机制,可响应应激而阻断细胞增殖。然而,近期证据表明,衰老的肿瘤细胞能够重新进入细胞周期,进而转变为癌症干细胞,导致癌症化疗后复发。了解衰老重编程过程如何成为癌症干细胞形成的前奏具有重大医学意义。为研究衰老、干性与癌症之间的相互作用,我们将干细胞培养基(SCM)应用于人类胚胎成纤维细胞(MRC5和WI-38)以及癌细胞系(A549和293T)。用SCM处理的MRC5和WI-38细胞出现氧化应激症状并发生衰老。对SCM诱导衰老的时间进程进行转录组分析,发现一个与生长停滞基因上调以及衰老相关分泌表型(SASP)重叠的发育过程。我们证明,通过重塑染色质结构发挥作用的组蛋白去甲基化酶含jumonji结构域蛋白D3(Jmjd3)和X染色体普遍转录四肽重复蛋白(Utx)参与衰老重编程过程,以阻断成纤维细胞中干细胞的形成。相比之下,在SCM中培养的A549和293T细胞转变为癌症干细胞,表现出与生长停滞解偶联的衰老表型。DNA甲基转移酶(Dnmt1和Dnmt3A)、十一-易位甲基胞嘧啶双加氧酶(Tet1和Tet3)、Jmjd3和Utx蛋白的直接过表达可激活293T细胞中的衰老相关β-半乳糖苷酶(SA-β-gal)活性,表明表观遗传改变和染色质重塑因子触发衰老反应。总体而言,我们的研究表明,控制衰老重编程的染色质机制在癌症干细胞形成中具有重要意义。