Santos Jaime, Pujols Jordi, Pallarès Irantzu, Iglesias Valentín, Ventura Salvador
Institut de Biotecnologia i Biomedicina and Departament de Bioquímica i Biologia Molecular, Universitat Autònoma de Barcelona, Bellaterra, Barcelona, Spain.
Comput Struct Biotechnol J. 2020 Jun 10;18:1403-1413. doi: 10.1016/j.csbj.2020.05.026. eCollection 2020.
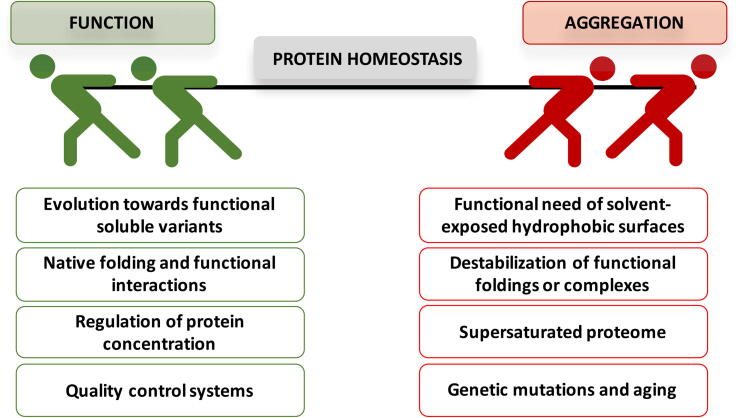
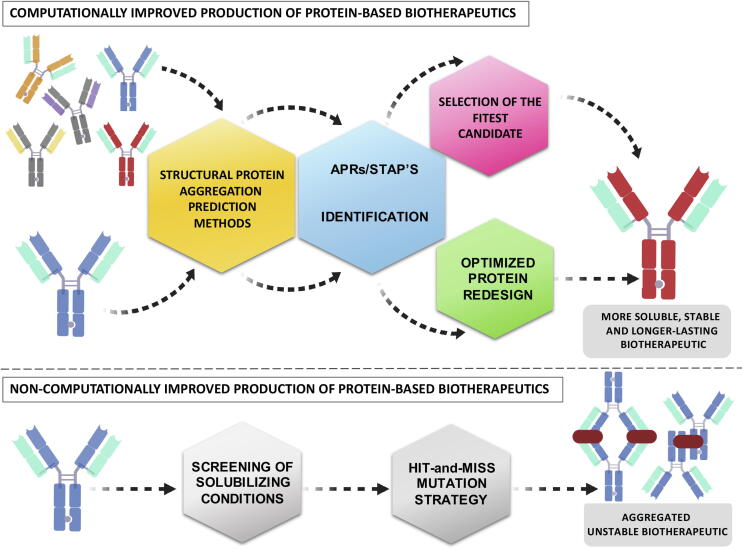
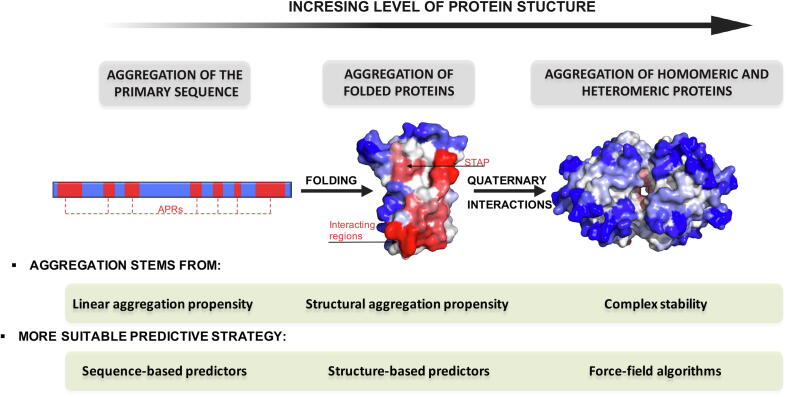
Protein aggregation is a widespread phenomenon that stems from the establishment of non-native intermolecular contacts resulting in protein precipitation. Despite its deleterious impact on fitness, protein aggregation is a generic property of polypeptide chains, indissociable from protein structure and function. Protein aggregation is behind the onset of neurodegenerative disorders and one of the serious obstacles in the production of protein-based therapeutics. The development of computational tools opened a new avenue to rationalize this phenomenon, enabling prediction of the aggregation propensity of individual proteins as well as proteome-wide analysis. These studies spotted aggregation as a major force driving protein evolution. Actual algorithms work on both protein sequences and structures, some of them accounting also for conformational fluctuations around the native state and the protein microenvironment. This toolbox allows to delineate conformation-specific routines to assist in the identification of aggregation-prone regions and to guide the optimization of more soluble and stable biotherapeutics. Here we review how the advent of predictive tools has change the way we think and address protein aggregation.
蛋白质聚集是一种普遍现象,它源于非天然分子间相互作用的建立,导致蛋白质沉淀。尽管蛋白质聚集对适应性有有害影响,但它是多肽链的一种普遍特性,与蛋白质的结构和功能密不可分。蛋白质聚集是神经退行性疾病发病的原因,也是基于蛋白质的治疗药物生产中的严重障碍之一。计算工具的发展为合理化这一现象开辟了新途径,能够预测单个蛋白质的聚集倾向以及全蛋白质组分析。这些研究发现聚集是驱动蛋白质进化的主要力量。实际算法可处理蛋白质序列和结构,其中一些算法还考虑了天然状态周围的构象波动和蛋白质微环境。这个工具箱有助于描绘特定构象的程序,以协助识别易聚集区域,并指导优化更易溶和稳定的生物治疗药物。在这里,我们回顾了预测工具的出现如何改变了我们思考和应对蛋白质聚集的方式。