Faculty of Medicine, Health and Human Sciences, Macquarie University, F10A, 2 Technology Place, North Ryde, NSW, 2109, Australia.
Australian Proteome Analysis Facility, Macquarie University, North Ryde, NSW, 2109, Australia.
J Transl Med. 2020 Jul 9;18(1):278. doi: 10.1186/s12967-020-02448-z.
Severe acute respiratory syndrome (SARS) has been initiating pandemics since the beginning of the century. In December 2019, the world was hit again by a devastating SARS episode that has so far infected almost four million individuals worldwide, with over 200,000 fatalities having already occurred by mid-April 2020, and the infection rate continues to grow exponentially. SARS coronavirus 2 (SARS-CoV-2) is a single stranded RNA pathogen which is characterised by a high mutation rate. It is vital to explore the mutagenic capability of the viral genome that enables SARS-CoV-2 to rapidly jump from one host immunity to another and adapt to the genetic pool of local populations.
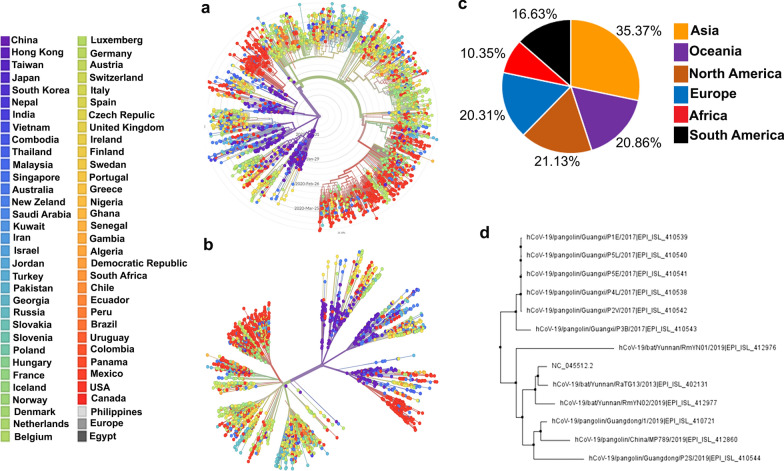
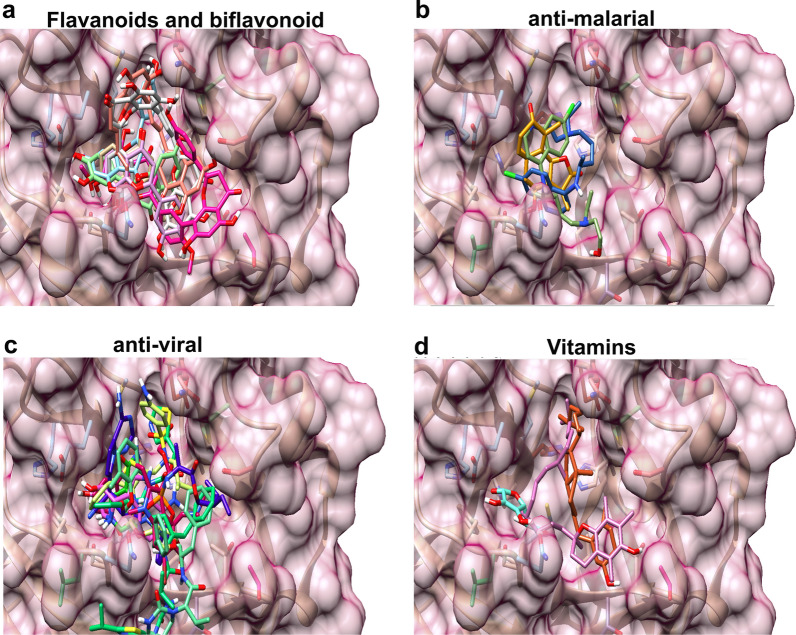
For this study, we analysed 2301 complete viral sequences reported from SARS-CoV-2 infected patients. SARS-CoV-2 host genomes were collected from The Global Initiative on Sharing All Influenza Data (GISAID) database containing 9 genomes from pangolin-CoV origin and 3 genomes from bat-CoV origin, Wuhan SARS-CoV2 reference genome was collected from GeneBank database. The Multiple sequence alignment tool, Clustal Omega was used for genomic sequence alignment. The viral replicating enzyme, 3-chymotrypsin-like cysteine protease (3CL) that plays a key role in its pathogenicity was used to assess its affinity with pharmacological inhibitors and repurposed drugs such as anti-viral flavones, biflavanoids, anti-malarial drugs and vitamin supplements.
Our results demonstrate that bat-CoV shares > 96% similar identity, while pangolin-CoV shares 85.98% identity with Wuhan SARS-CoV-2 genome. This in-depth analysis has identified 12 novel recurrent mutations in South American and African viral genomes out of which 3 were unique in South America, 4 unique in Africa and 5 were present in-patient isolates from both populations. Using state of the art in silico approaches, this study further investigates the interaction of repurposed drugs with the SARS-CoV-2 3CL enzyme, which regulates viral replication machinery.
Overall, this study provides insights into the evolving mutations, with implications to understand viral pathogenicity and possible new strategies for repurposing compounds to combat the nCovid-19 pandemic.
严重急性呼吸系统综合症(SARS)自本世纪初以来引发了多次大流行。2019 年 12 月,世界再次遭受了一场毁灭性的 SARS 疫情的打击,迄今为止,全球已有近 400 万人感染,截至 2020 年 4 月中旬,已有超过 20 万人死亡,感染率仍在呈指数级增长。SARS 冠状病毒 2(SARS-CoV-2)是一种单链 RNA 病原体,其特点是突变率很高。探索病毒基因组的诱变能力至关重要,这使 SARS-CoV-2 能够迅速从一个宿主免疫跳到另一个宿主免疫,并适应当地人群的遗传库。
在这项研究中,我们分析了来自 SARS-CoV-2 感染患者的 2301 个完整病毒序列。从 The Global Initiative on Sharing All Influenza Data(GISAID)数据库中收集 SARS-CoV-2 宿主基因组,该数据库包含 9 个来自穿山甲-CoV 的基因组和 3 个来自蝙蝠-CoV 的基因组,从 GeneBank 数据库中收集武汉 SARS-CoV2 参考基因组。使用多重序列比对工具 Clustal Omega 进行基因组序列比对。3-胰凝乳蛋白酶样半胱氨酸蛋白酶(3CL)是一种在其致病性中起关键作用的病毒复制酶,用于评估其与药理学抑制剂和再利用药物(如抗病毒黄酮类、双黄酮类、抗疟药物和维生素补充剂)的亲和力。
我们的结果表明,蝙蝠-CoV 与武汉 SARS-CoV-2 基因组的相似度超过 96%,而穿山甲-CoV 的相似度为 85.98%。这项深入分析在南美和非洲病毒基因组中发现了 12 个新的反复出现的突变,其中 3 个是南美特有的,4 个是非洲特有的,5 个是来自这两个地区的患者分离株。本研究利用最先进的计算方法,进一步研究了再利用药物与调节病毒复制机制的 SARS-CoV-2 3CL 酶的相互作用。
总的来说,这项研究提供了对不断进化的突变的深入了解,有助于了解病毒的致病性,并为对抗 nCovid-19 大流行寻找重新利用化合物的新策略提供了依据。