Hotokezaka Yuka, Katayama Ikuo, Nakamura Takashi
Department of Radiology and Cancer Biology, Nagasaki University Graduate School of Biomedical Sciences 1-7-1 Sakamoto, Nagasaki, 852-8588, Japan.
Commun Biol. 2020 Jul 14;3(1):378. doi: 10.1038/s42003-020-1102-2.
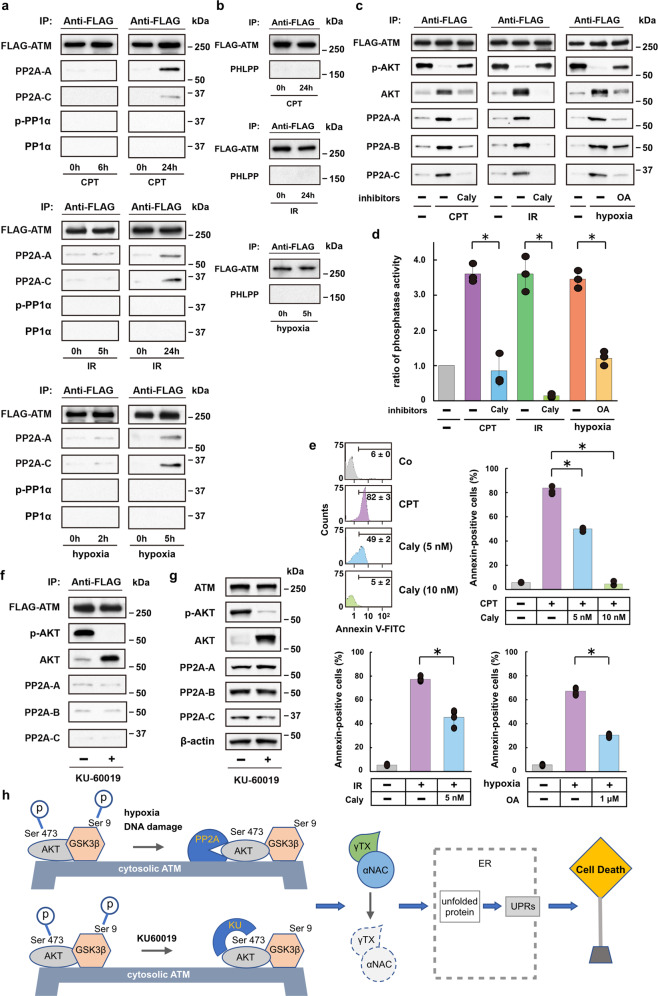
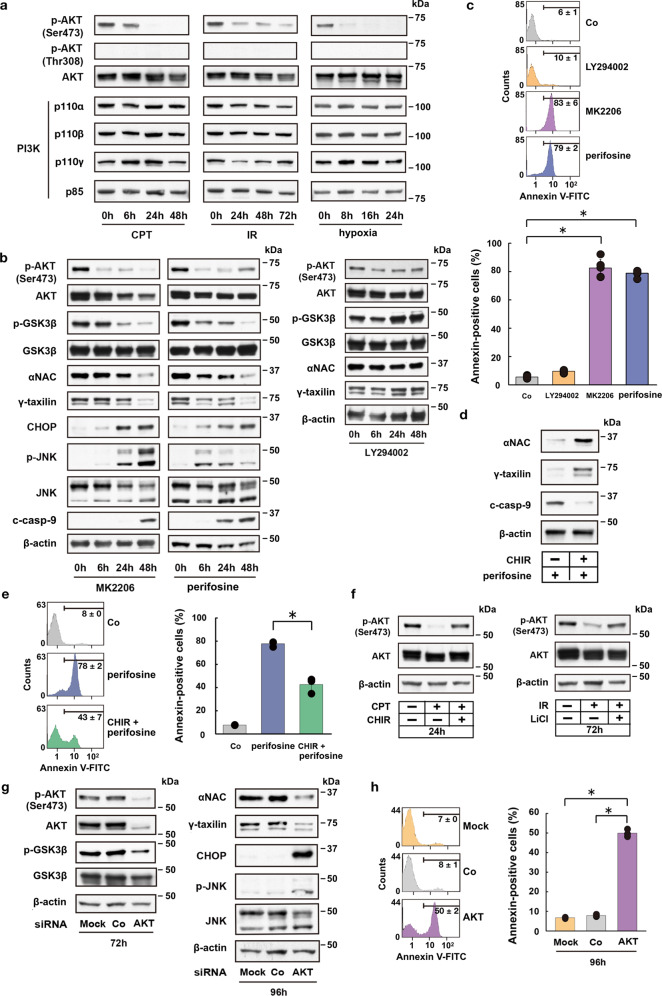
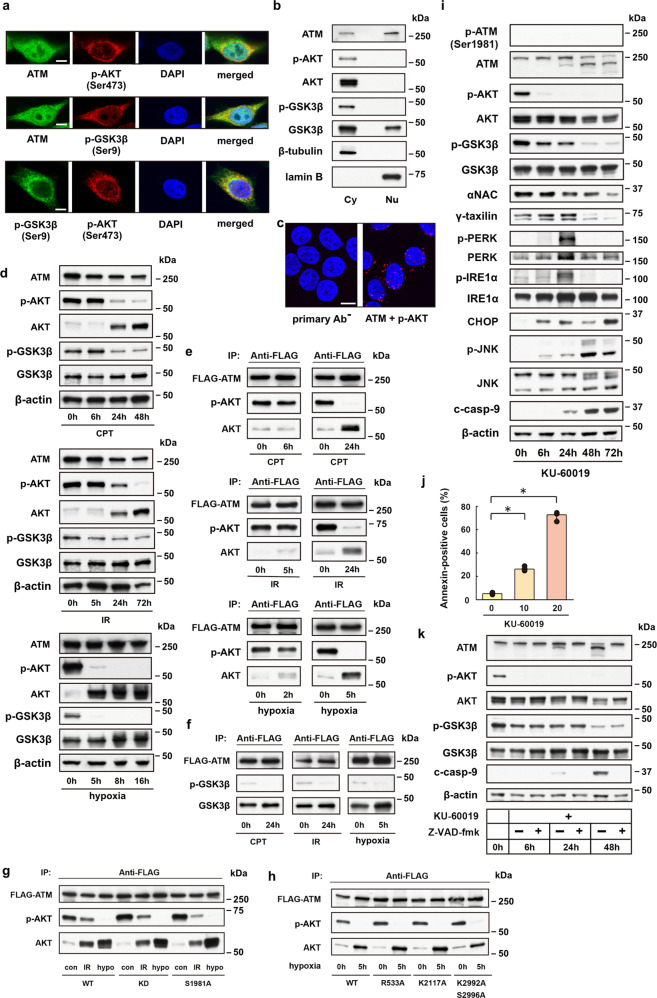
Endoplasmic reticulum (ER) stress can be caused by perturbations in ER function resulting from the accumulation of unfolded/misfolded proteins in the ER lumen. Accumulating unfolded proteins trigger unfolded protein responses (UPRs) through activating three transmembrane sensors on the ER: IRE1α, PERK, and ATF6. The orchestrated action of these molecules upregulates genes encoding proteins involved in the downregulation of protein synthesis and acceleration of protein secretion. Ineffectiveness of these fail-safe mechanisms may lead to apoptosis. However, the molecular mechanisms upstream of the UPR are not fully understood. Here we show participation of ataxia telangiectasia mutated (ATM) in stress-induced apoptosis. Cytoplasmic ATM serves as a platform on which protein phosphatase 2A-dependent dephosphorylation of AKT activates glycogen synthase kinase 3β, thereby downregulating nascent polypeptide-associated complex α subunit and γ-taxilin, triggering UPRs and leading to mitochondria-dependent apoptosis. These results suggest an ATM/AKT-dependent cell death pathway triggered by various forms of stress.
内质网(ER)应激可由内质网腔中未折叠/错误折叠蛋白质的积累导致内质网功能紊乱引起。积累的未折叠蛋白质通过激活内质网上的三种跨膜传感器:肌醇需求酶1α(IRE1α)、蛋白激酶R样内质网激酶(PERK)和活化转录因子6(ATF6)来触发未折叠蛋白反应(UPR)。这些分子的协同作用上调了编码参与蛋白质合成下调和蛋白质分泌加速的蛋白质的基因。这些故障安全机制的失效可能导致细胞凋亡。然而,UPR上游的分子机制尚未完全了解。在这里,我们展示了共济失调毛细血管扩张症突变基因(ATM)参与应激诱导的细胞凋亡。细胞质中的ATM作为一个平台,蛋白磷酸酶2A依赖的AKT去磷酸化在此平台上激活糖原合酶激酶3β,从而下调新生多肽相关复合物α亚基和γ-微管蛋白结合蛋白,触发UPR并导致线粒体依赖性细胞凋亡。这些结果表明存在由各种形式的应激触发的ATM/AKT依赖性细胞死亡途径。