Savage Sara R, Zhang Bing
Department of Biomedical Informatics, Vanderbilt University, Nashville, TN USA.
Lester and Sue Smith Breast Center, Baylor College of Medicine, Houston, TX USA.
Clin Proteomics. 2020 Jul 11;17:27. doi: 10.1186/s12014-020-09290-x. eCollection 2020.
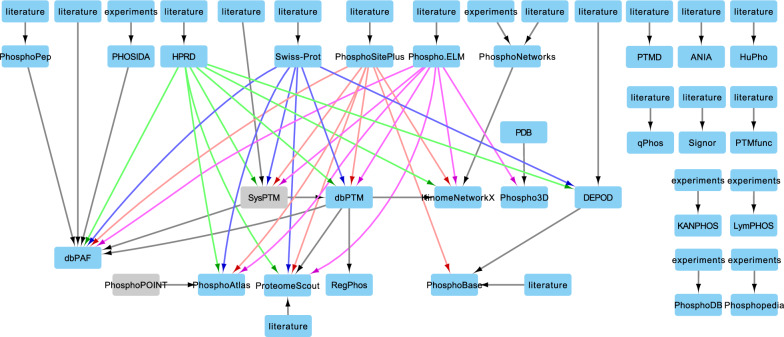
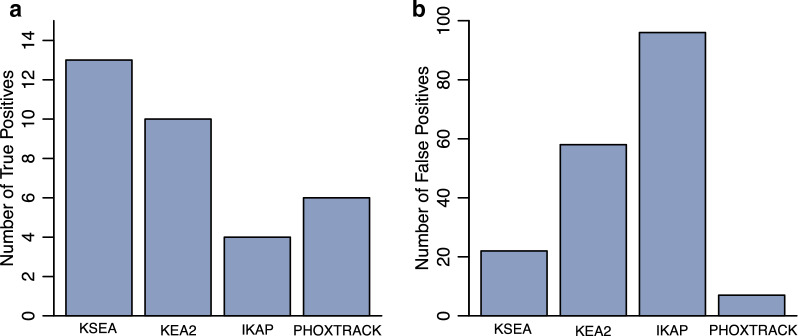
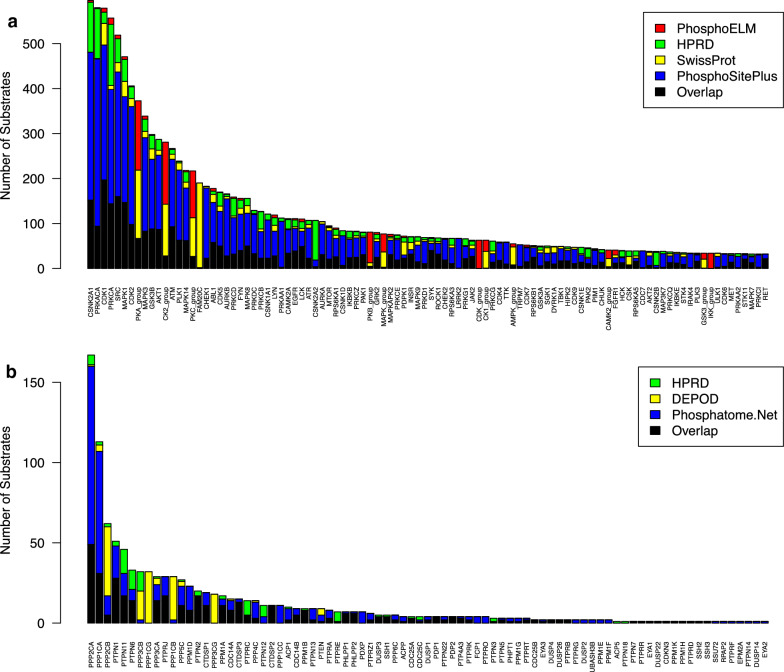
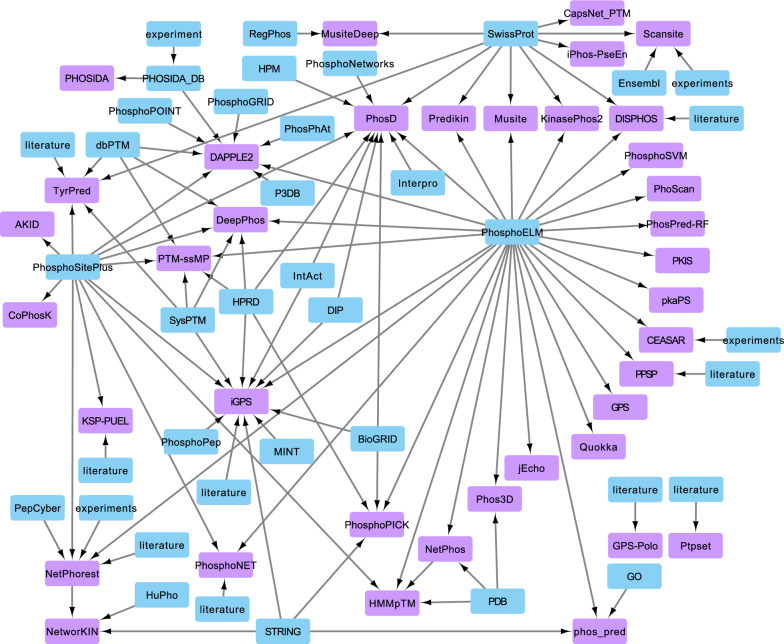
Mass spectrometry-based phosphoproteomics is becoming an essential methodology for the study of global cellular signaling. Numerous bioinformatics resources are available to facilitate the translation of phosphopeptide identification and quantification results into novel biological and clinical insights, a critical step in phosphoproteomics data analysis. These resources include knowledge bases of kinases and phosphatases, phosphorylation sites, kinase inhibitors, and sequence variants affecting kinase function, and bioinformatics tools that can predict phosphorylation sites in addition to the kinase that phosphorylates them, infer kinase activity, and predict the effect of mutations on kinase signaling. However, these resources exist in silos and it is challenging to select among multiple resources with similar functions. Therefore, we put together a comprehensive collection of resources related to phosphoproteomics data interpretation, compared the use of tools with similar functions, and assessed the usability from the standpoint of typical biologists or clinicians. Overall, tools could be improved by standardization of enzyme names, flexibility of data input and output format, consistent maintenance, and detailed manuals.
基于质谱的磷酸化蛋白质组学正成为研究全球细胞信号传导的重要方法。有许多生物信息学资源可用于促进将磷酸肽鉴定和定量结果转化为新的生物学和临床见解,这是磷酸化蛋白质组学数据分析中的关键步骤。这些资源包括激酶和磷酸酶、磷酸化位点、激酶抑制剂以及影响激酶功能的序列变体的知识库,以及除了能够磷酸化它们的激酶之外还可以预测磷酸化位点、推断激酶活性并预测突变对激酶信号传导影响的生物信息学工具。然而,这些资源分散存在,在多个功能相似的资源中进行选择具有挑战性。因此,我们汇集了与磷酸化蛋白质组学数据解释相关的综合资源集,比较了具有相似功能的工具的使用情况,并从典型生物学家或临床医生的角度评估了可用性。总体而言,工具可以通过酶名称的标准化、数据输入和输出格式的灵活性、持续一致的维护以及详细的手册来改进。