Istituto Auxologico Italiano, IRCCS, Center for Cardiac Arrhythmias of Genetic Origin and Laboratory of Cardiovascular Genetics, Milan, Italy.
European Reference Network for Rare and Low Prevalence Complex Diseases of the Heart (ERN GUARD-HEART), Bruxelles, Belgium.
Nat Rev Dis Primers. 2020 Jul 16;6(1):58. doi: 10.1038/s41572-020-0188-7.
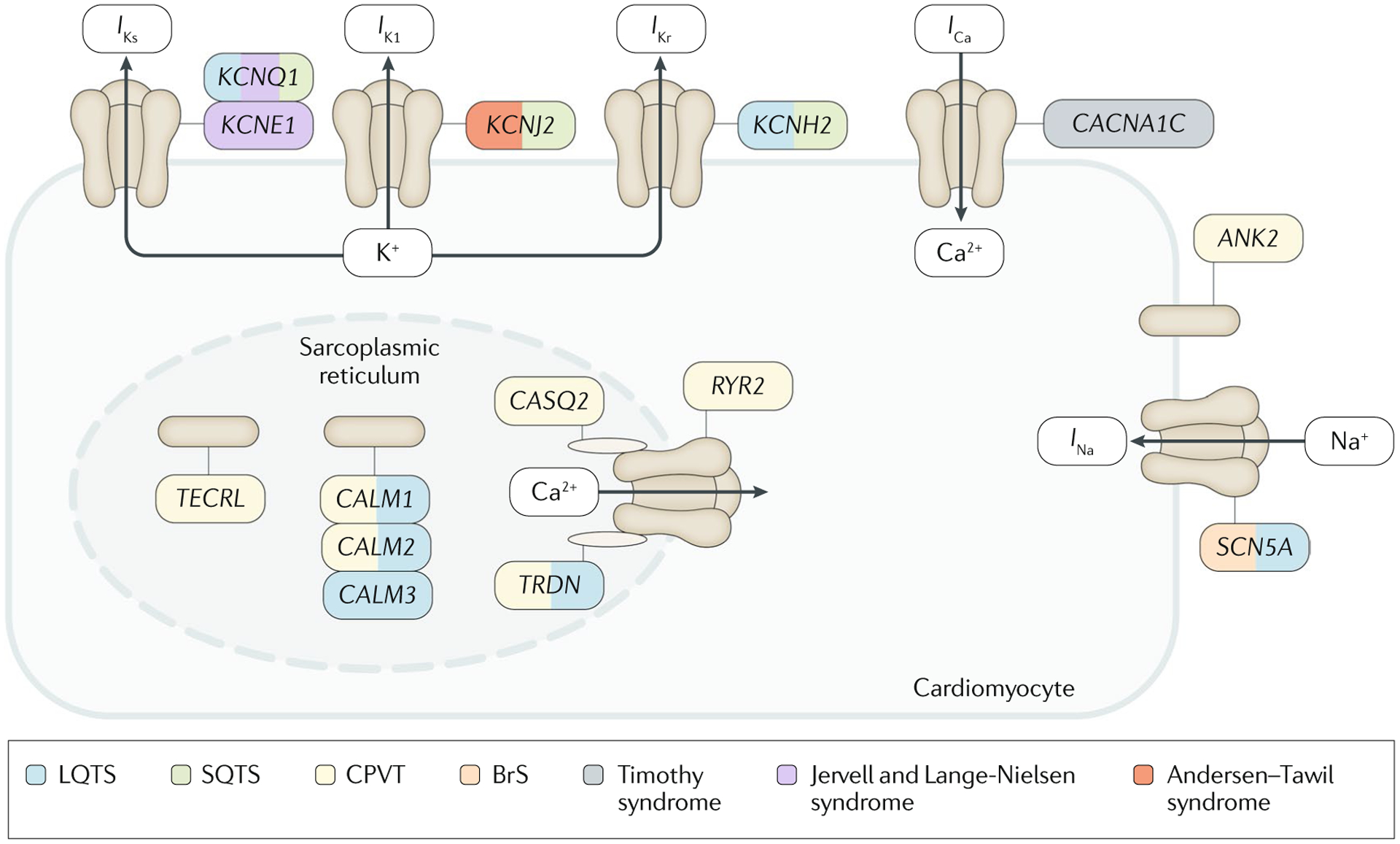
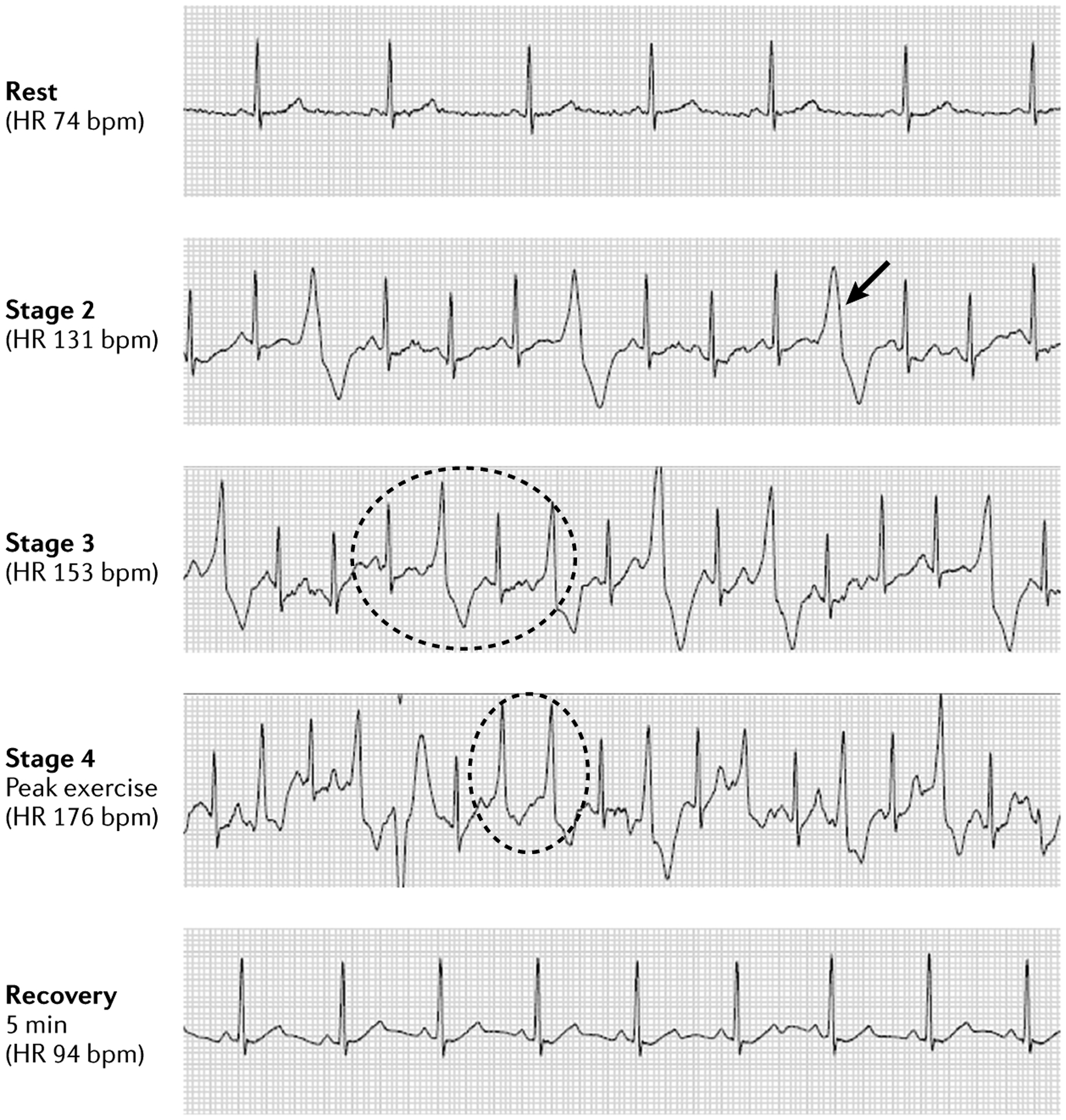
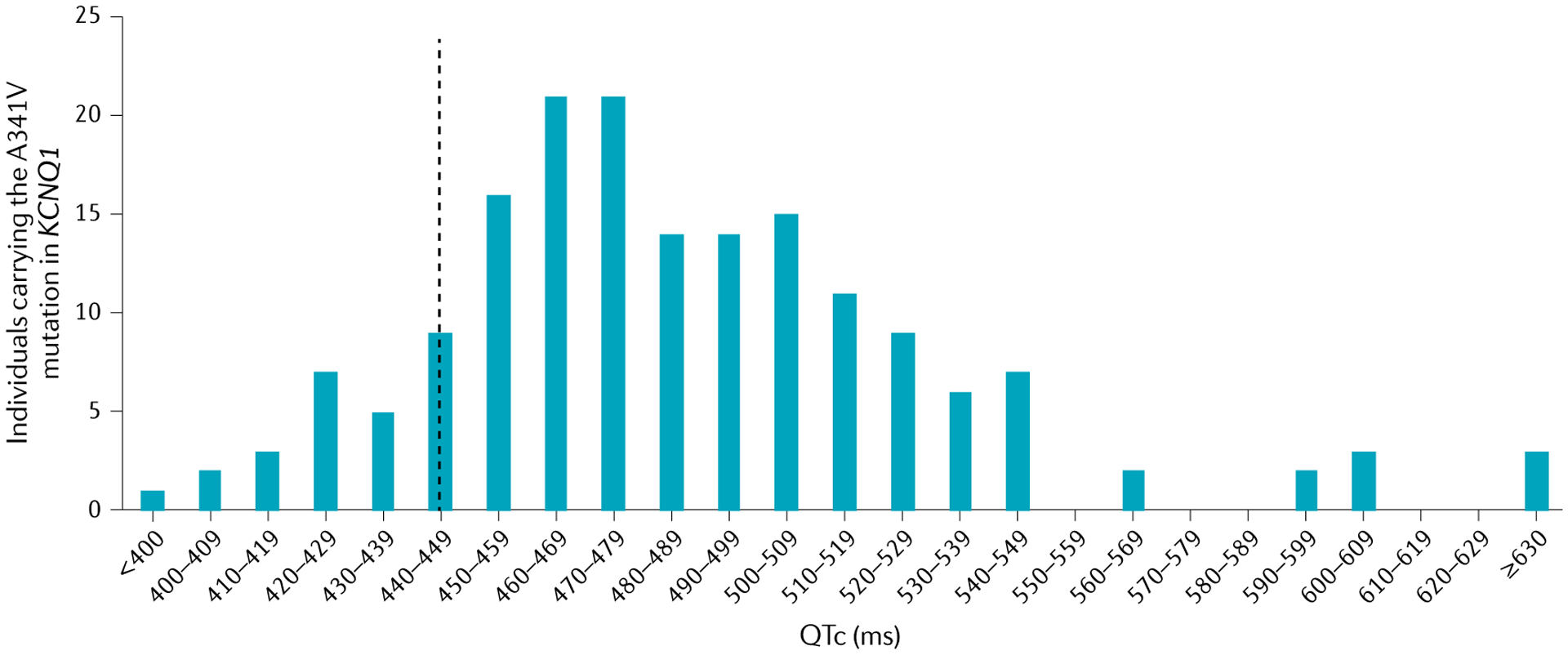
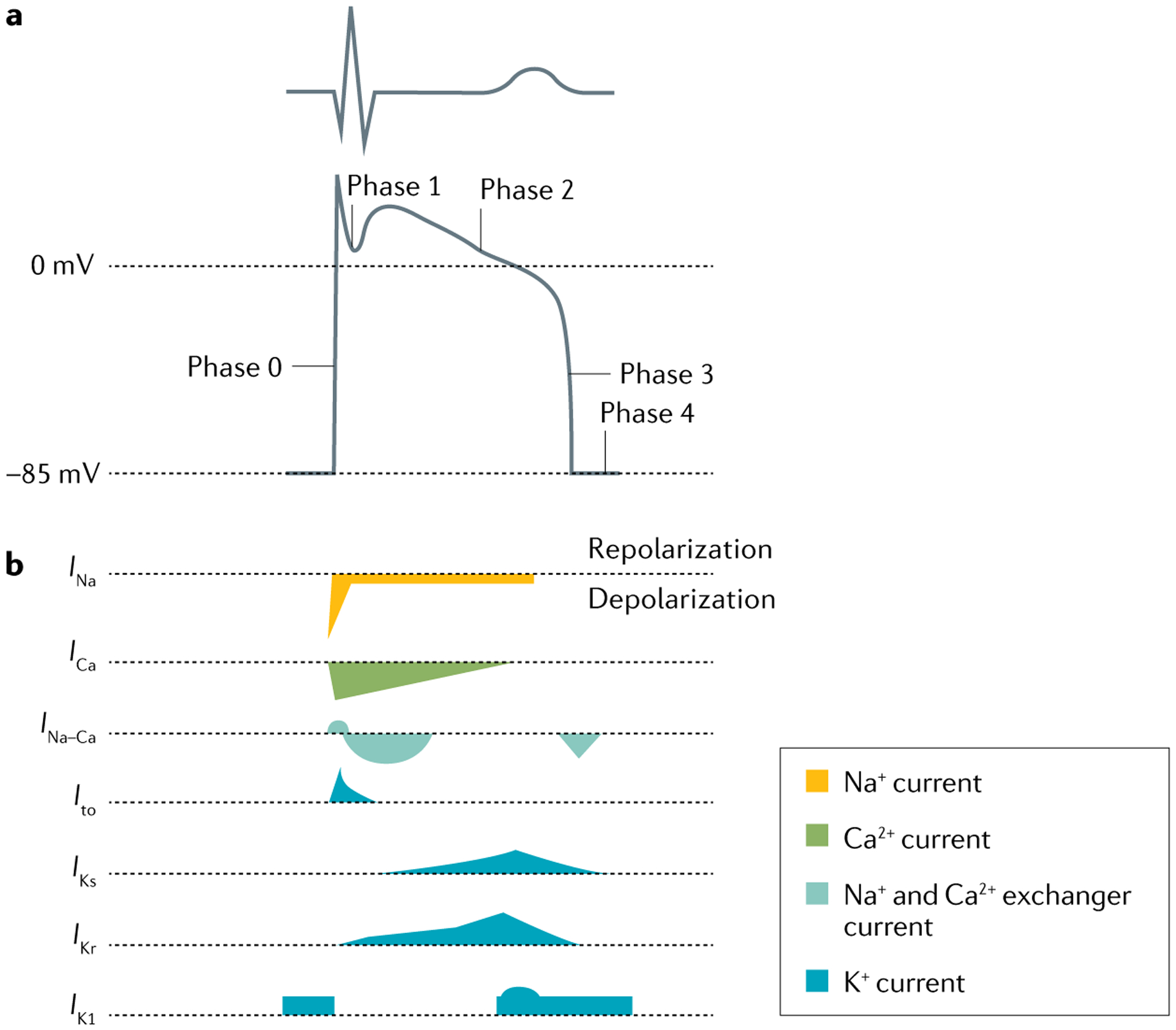
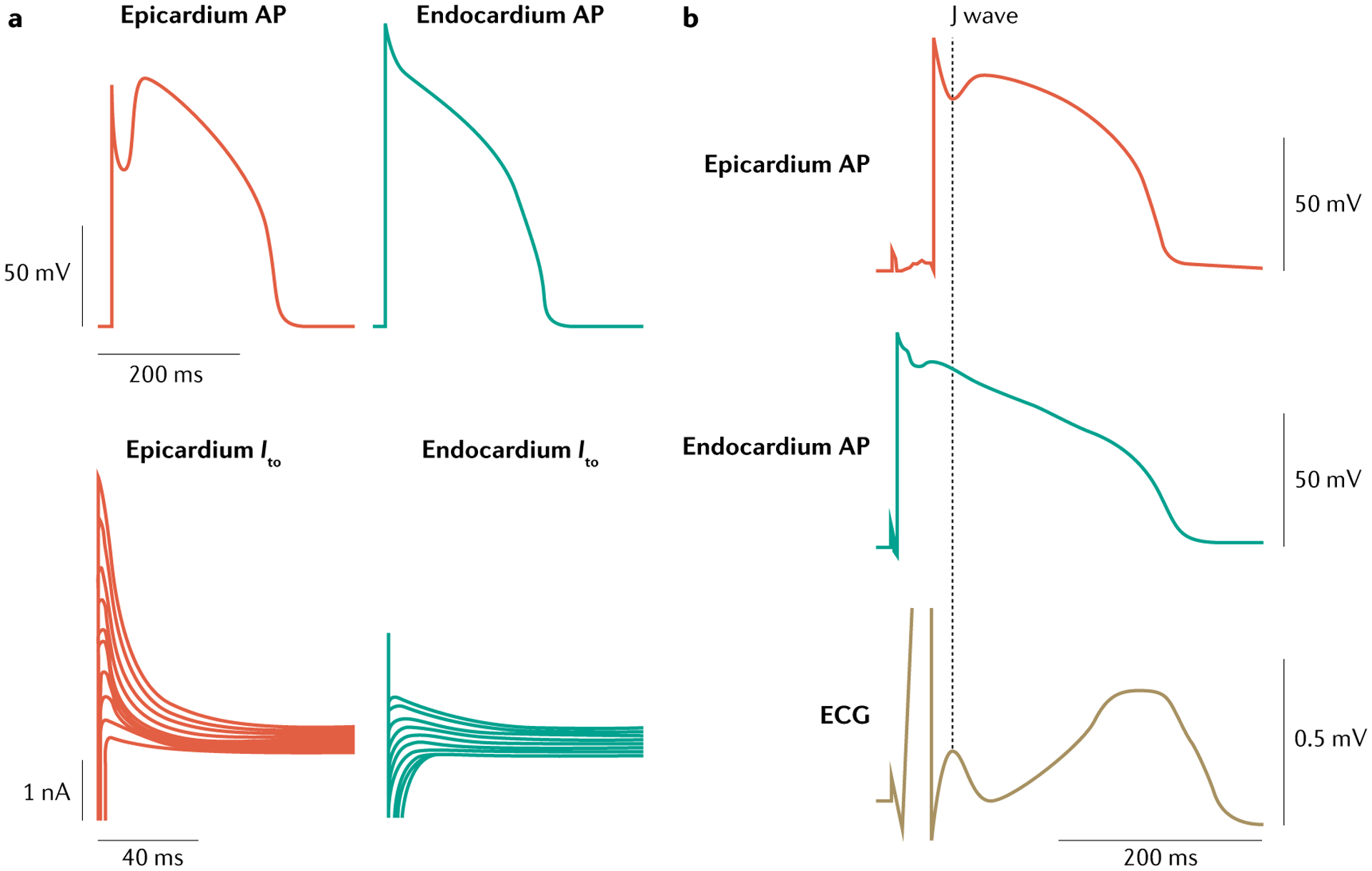
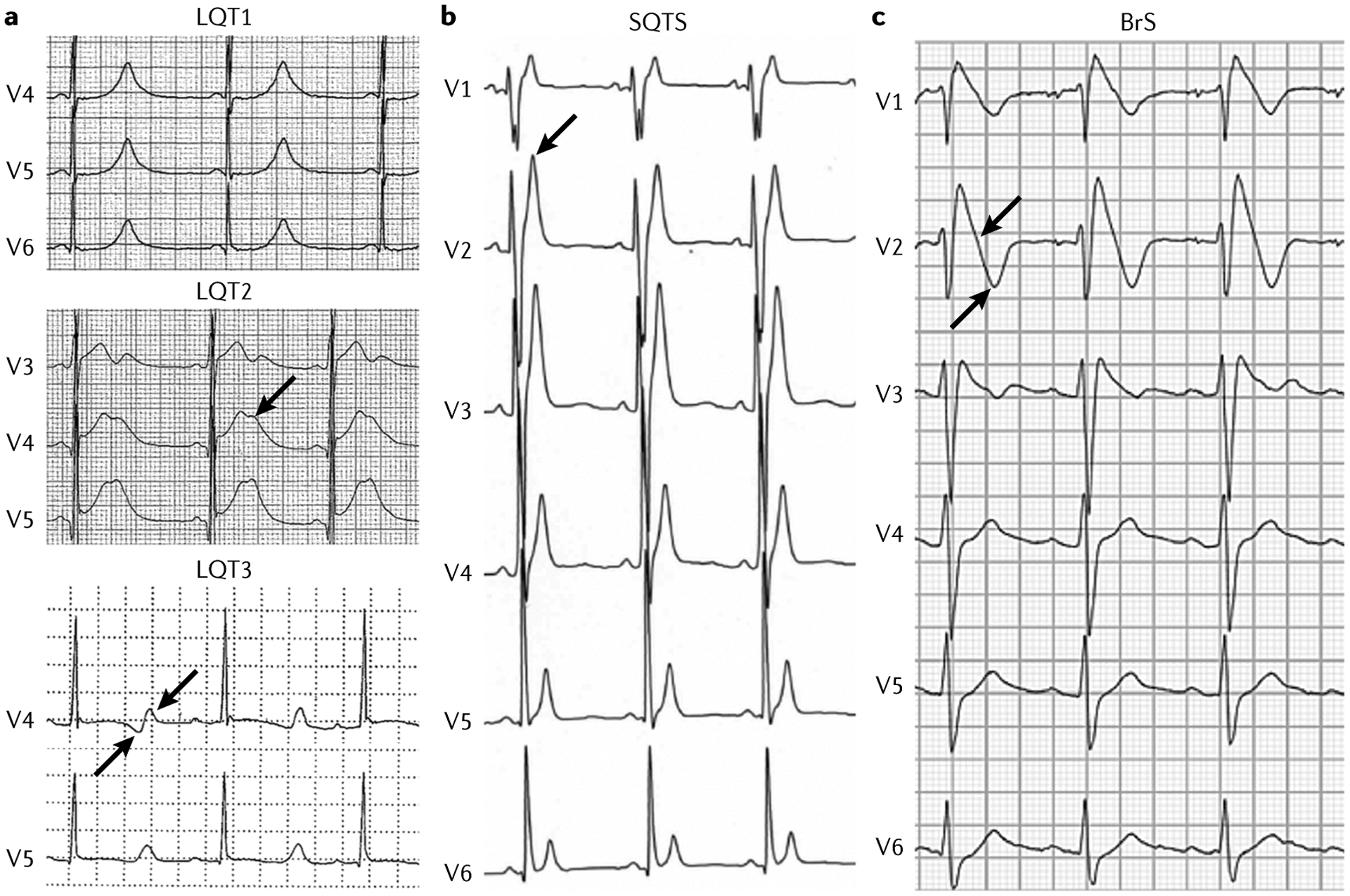
The main inherited cardiac arrhythmias are long QT syndrome, short QT syndrome, catecholaminergic polymorphic ventricular tachycardia and Brugada syndrome. These rare diseases are often the underlying cause of sudden cardiac death in young individuals and result from mutations in several genes encoding ion channels or proteins involved in their regulation. The genetic defects lead to alterations in the ionic currents that determine the morphology and duration of the cardiac action potential, and individuals with these disorders often present with syncope or a life-threatening arrhythmic episode. The diagnosis is based on clinical presentation and history, the characteristics of the electrocardiographic recording at rest and during exercise and genetic analyses. Management relies on pharmacological therapy, mostly β-adrenergic receptor blockers (specifically, propranolol and nadolol) and sodium and transient outward current blockers (such as quinidine), or surgical interventions, including left cardiac sympathetic denervation and implantation of a cardioverter-defibrillator. All these arrhythmias are potentially life-threatening and have substantial negative effects on the quality of life of patients. Future research should focus on the identification of genes associated with the diseases and other risk factors, improved risk stratification and, in particular for Brugada syndrome, effective therapies.
主要的遗传性心律失常包括长 QT 综合征、短 QT 综合征、儿茶酚胺多形性室性心动过速和 Brugada 综合征。这些罕见疾病常是年轻人心源性猝死的潜在病因,是由编码离子通道或参与其调节的蛋白的几个基因突变引起的。遗传缺陷导致决定心脏动作电位形态和持续时间的离子电流改变,这些疾病患者常表现为晕厥或危及生命的心律失常发作。诊断基于临床表现和病史、静息和运动时心电图记录的特征以及基因分析。治疗依赖于药物治疗,主要是β肾上腺素能受体阻滞剂(具体为普萘洛尔和纳多洛尔)和钠及瞬间外向电流阻滞剂(如奎尼丁),或手术干预,包括左心交感神经切断术和植入心脏除颤器。所有这些心律失常都有潜在的生命威胁,并对患者的生活质量产生重大负面影响。未来的研究应集中于识别与这些疾病和其他风险因素相关的基因,改善风险分层,特别是对 Brugada 综合征,应寻找有效的治疗方法。