Department of Computer Science and Engineering, Bangladesh University of Engineering and Technology, Dhaka, 1205, Bangladesh.
Department of Computer Science and Engineering, Eastern University, Dhaka, Bangladesh.
BMC Genomics. 2020 Jul 20;21(1):497. doi: 10.1186/s12864-020-06892-5.
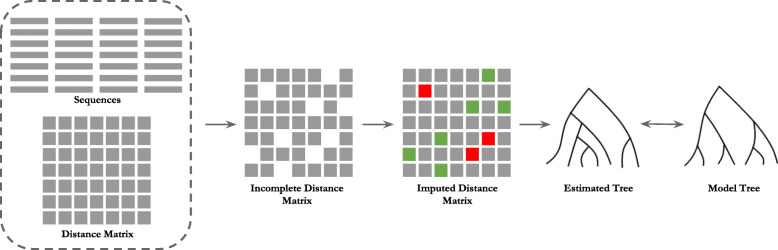
With the rapid growth rate of newly sequenced genomes, species tree inference from genes sampled throughout the whole genome has become a basic task in comparative and evolutionary biology. However, substantial challenges remain in leveraging these large scale molecular data. One of the foremost challenges is to develop efficient methods that can handle missing data. Popular distance-based methods, such as NJ (neighbor joining) and UPGMA (unweighted pair group method with arithmetic mean) require complete distance matrices without any missing data.
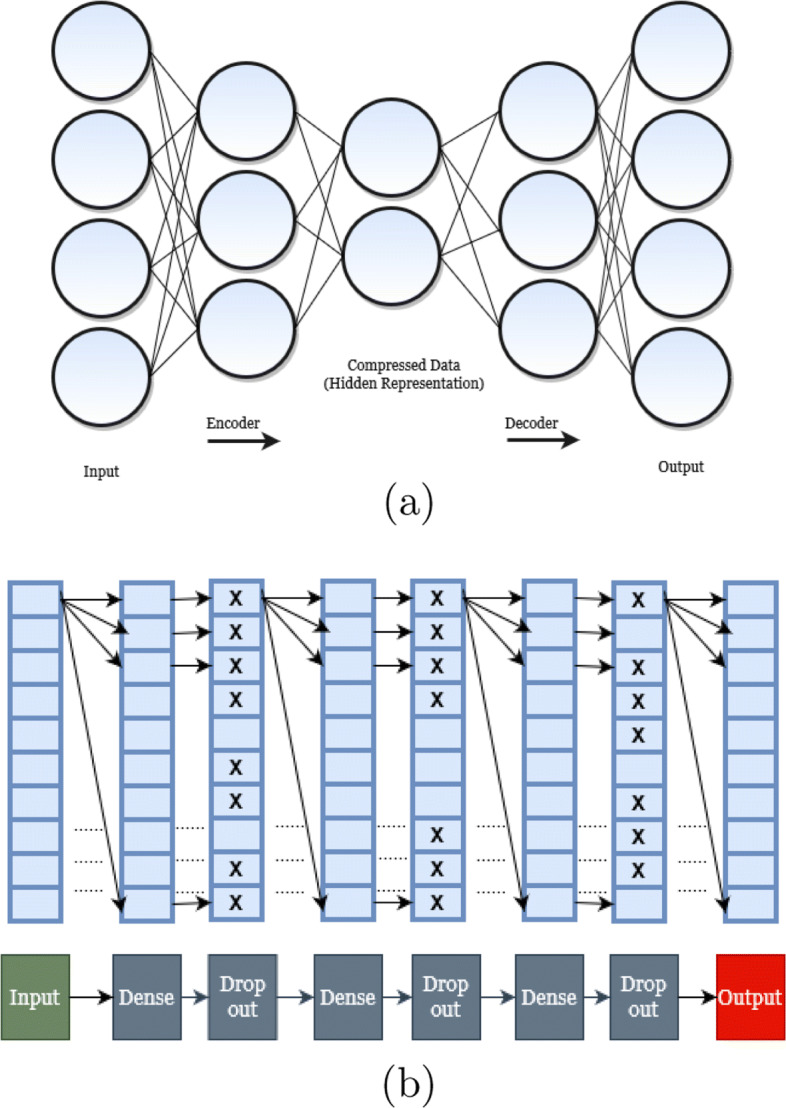
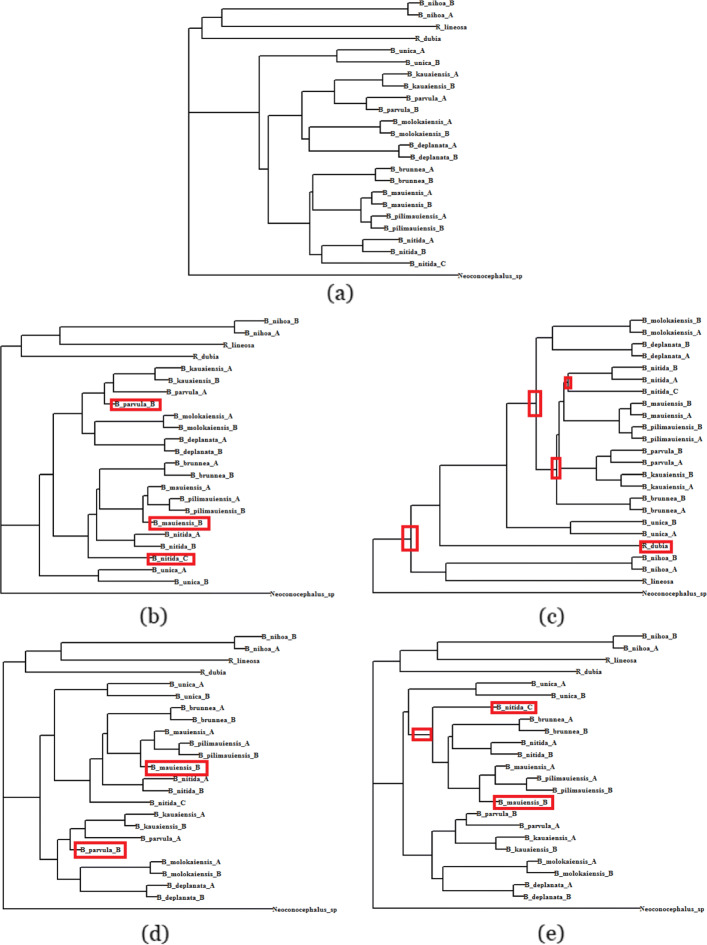
We introduce two highly accurate machine learning based distance imputation techniques. These methods are based on matrix factorization and autoencoder based deep learning architectures. We evaluated these two methods on a collection of simulated and biological datasets. Experimental results suggest that our proposed methods match or improve upon the best alternate distance imputation techniques. Moreover, these methods are scalable to large datasets with hundreds of taxa, and can handle a substantial amount of missing data.
This study shows, for the first time, the power and feasibility of applying deep learning techniques for imputing distance matrices. Thus, this study advances the state-of-the-art in phylogenetic tree construction in the presence of missing data. The proposed methods are available in open source form at https://github.com/Ananya-Bhattacharjee/ImputeDistances .
随着新测序基因组的快速增长,从整个基因组中采样的基因推断种系进化树已成为比较和进化生物学的基本任务。然而,在利用这些大规模分子数据方面仍然存在重大挑战。其中最主要的挑战之一是开发能够处理缺失数据的有效方法。流行的基于距离的方法,如 NJ(邻接法)和 UPGMA(算术平均未加权对组法),需要没有任何缺失数据的完整距离矩阵。
我们引入了两种基于机器学习的高度准确的距离填补技术。这些方法基于矩阵分解和基于自动编码器的深度学习架构。我们在一系列模拟和生物数据集上评估了这两种方法。实验结果表明,我们提出的方法与最佳替代距离填补技术相匹配或有所改进。此外,这些方法可扩展到具有数百个分类单元的大型数据集,并可以处理大量缺失数据。
这项研究首次展示了应用深度学习技术填补距离矩阵的强大功能和可行性。因此,这项研究在存在缺失数据的情况下推进了系统发育树构建的最新技术。所提出的方法可在 https://github.com/Ananya-Bhattacharjee/ImputeDistances 上以开源形式获得。