Kozak Rouba, Kiss Tamás, Dlugolenski Keith, Johnson David E, Gorczyca Roxanne R, Kuszpit Kyle, Harvey Brian D, Stolyar Polina, Sukoff Rizzo Stacey J, Hoffmann William E, Volfson Dmitri, Hajós Mihaly, Davoren Jennifer E, Abbott Amanda L, Williams Graham V, Castner Stacy A, Gray David L
Global Research and Development, Pfizer Inc., Groton, CT, United States.
Department of Comparative Medicine, Yale School of Medicine, New Haven, CT, United States.
Front Pharmacol. 2020 Jul 7;11:1005. doi: 10.3389/fphar.2020.01005. eCollection 2020.
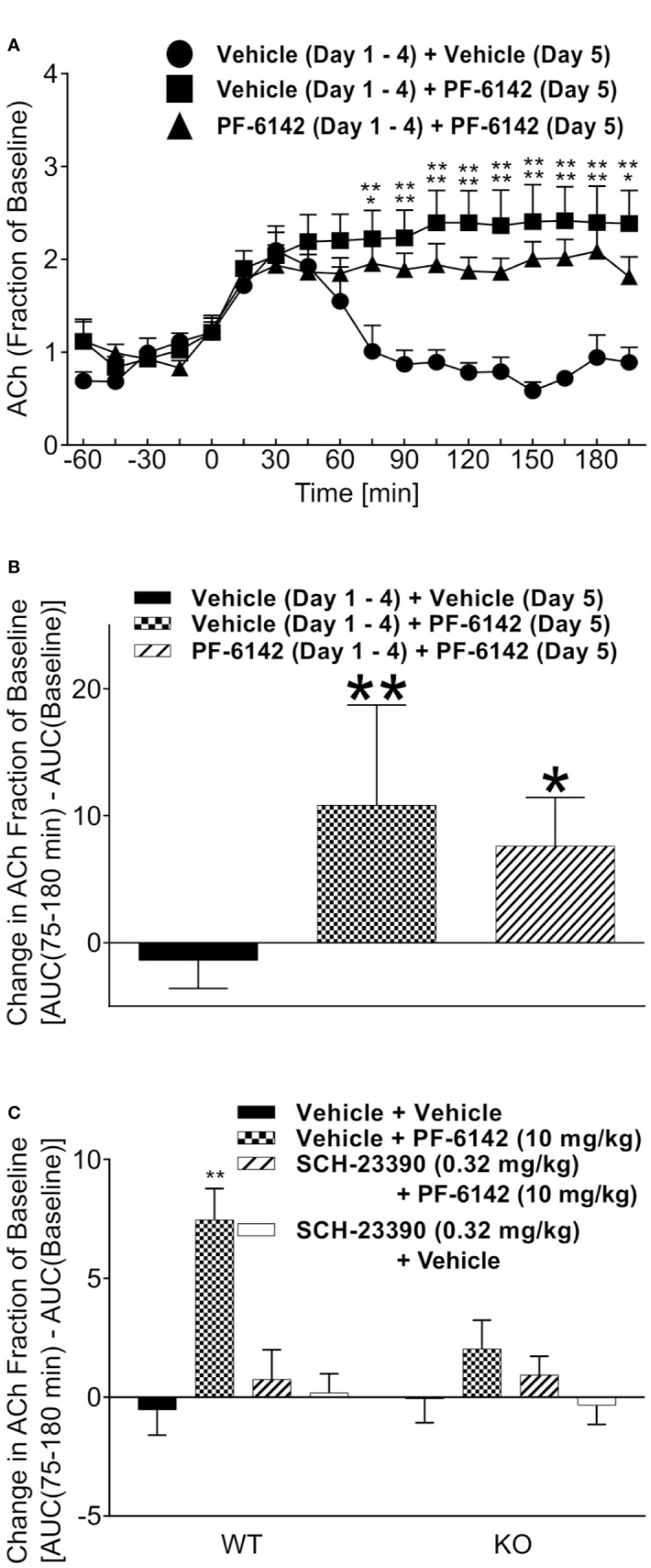
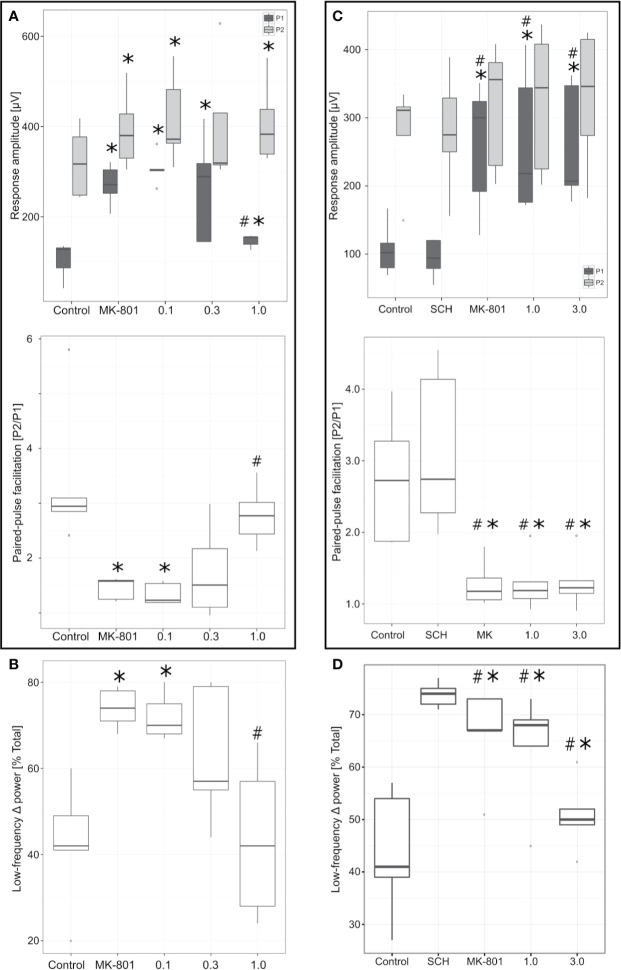
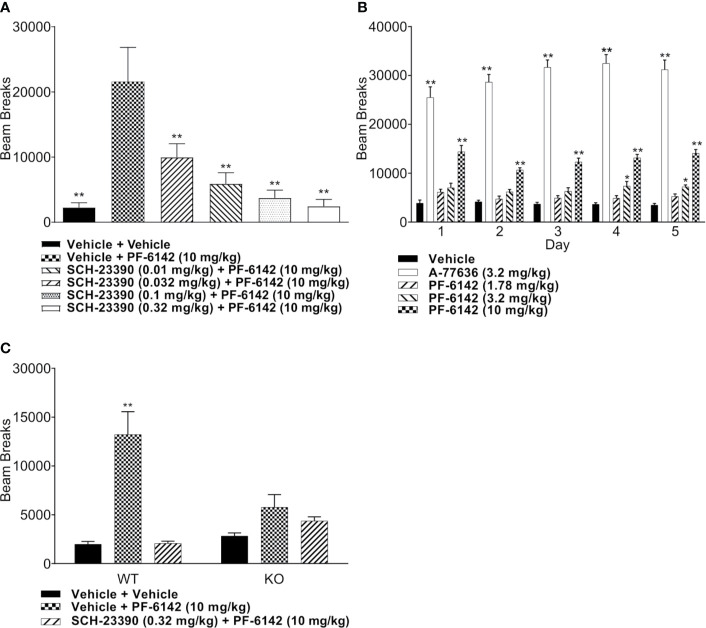
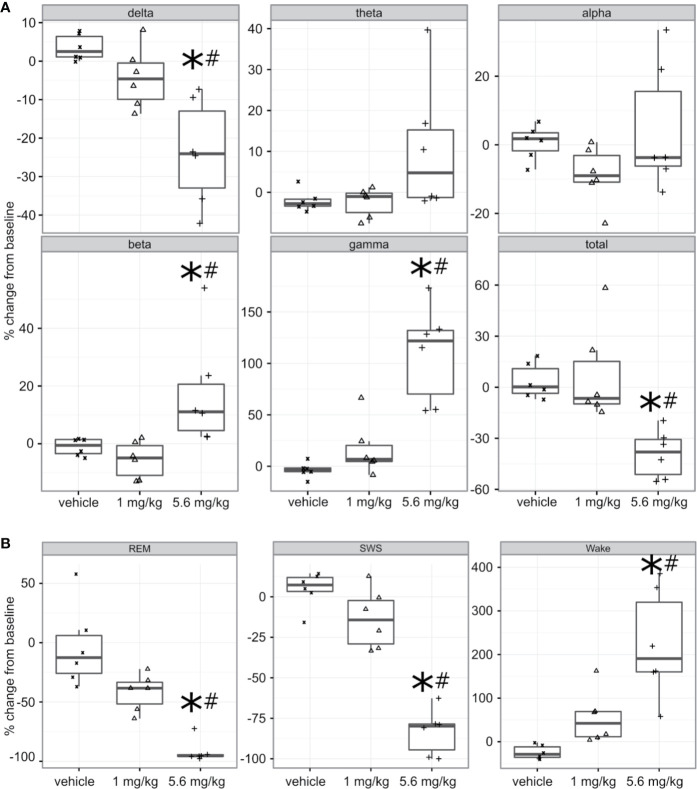
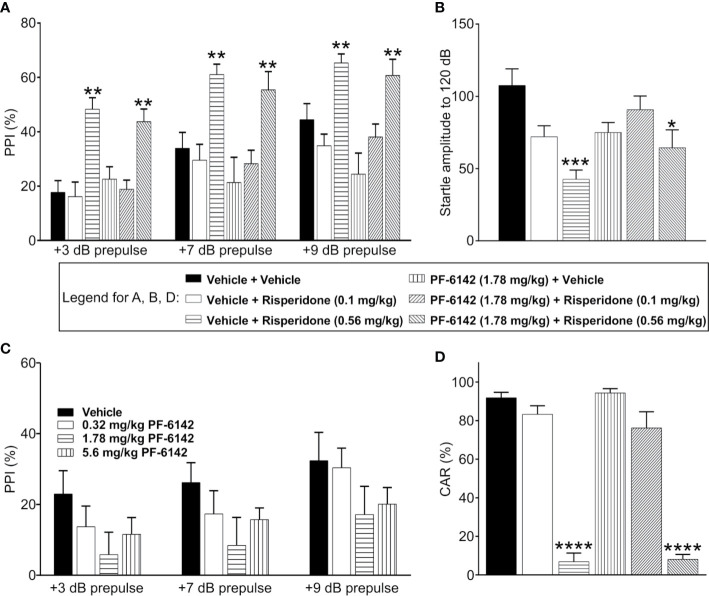
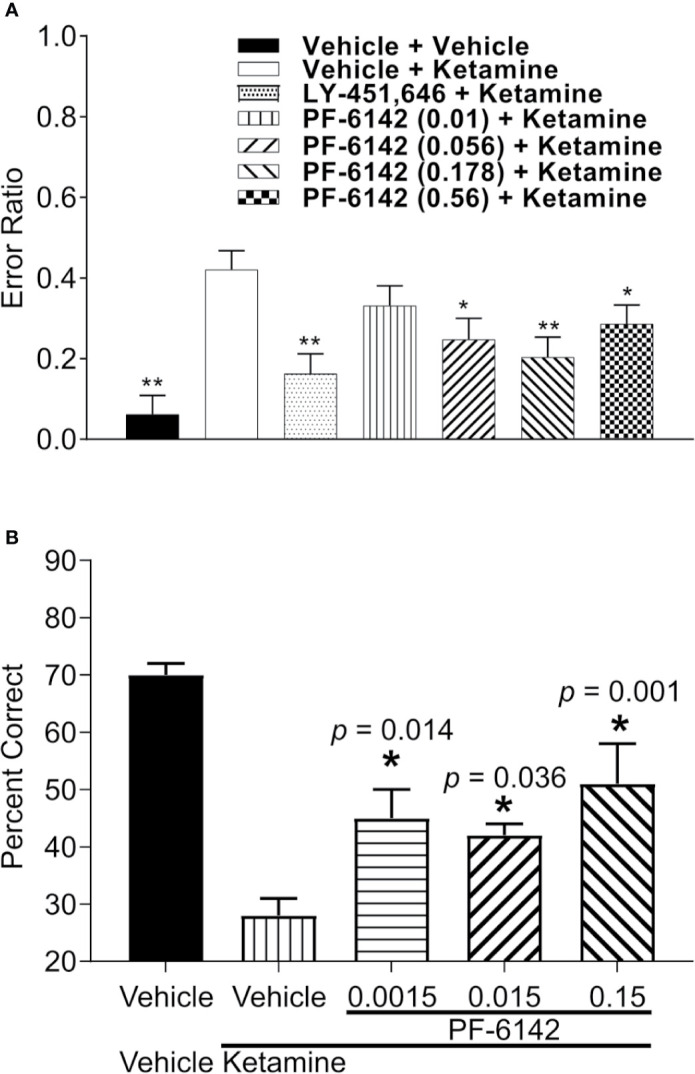
Selective activation of dopamine D1 receptors remains a promising pro-cognitive therapeutic strategy awaiting robust clinical investigation. PF-6142 is a key example from a recently disclosed novel series of non-catechol agonists and partial agonists of the dopamine D1/5 receptors (D1R) that exhibit pharmacokinetic (PK) properties suitable for oral delivery. Given their reported potential for functionally biased signaling compared to known catechol-based selective agonists, and the promising rodent PK profile of PF-6142, we utilized relevant assays in male rodents and male and female non-human primates (NHP) to evaluate the pharmacology of this new series. Studies in rodents showed that PF-6142 increased locomotor activity and prefrontal cortex acetylcholine release, increased time spent in wakefulness, and desynchronized the EEG, like known D1R agonists. D1R selectivity of PF-6142 was supported by lack of effect in D1R knock-out mice and blocked response in the presence of the D1R antagonist SCH-23390. Further, PF-6142 improved performance in rodent models of NMDA receptor antagonist-induced cognitive dysfunction, such as MK-801-disrupted paired-pulse facilitation, and ketamine-disrupted working memory performance in the radial arm maze. Similarly, PF-6142 reversed ketamine-induced deficits in NHP performing the spatial delayed recognition task. Of importance, PF-6142 did not alter the efficacy of risperidone in assays predictive of antipsychotic-like effect in rodents including pre-pulse inhibition and conditioned avoidance responding. These data support the continued development of non-catechol based D1R agonists for the treatment of cognitive impairment associated with brain disorders including schizophrenia.
选择性激活多巴胺D1受体仍然是一种有前景的促认知治疗策略,有待进行深入的临床研究。PF-6142是最近披露的一系列新型非儿茶酚胺类多巴胺D1/5受体(D1R)激动剂和部分激动剂中的关键实例,其展现出适合口服给药的药代动力学(PK)特性。鉴于与已知的基于儿茶酚胺的选择性激动剂相比,它们具有功能偏向性信号传导的潜在特性,以及PF-6142在啮齿动物中展现出的有前景的PK特征,我们利用雄性啮齿动物以及雄性和雌性非人灵长类动物(NHP)的相关试验来评估这一新系列药物的药理学特性。啮齿动物研究表明,PF-6142可增加运动活性和前额叶皮层乙酰胆碱释放,增加清醒时间,并使脑电图去同步化,类似于已知的D1R激动剂。PF-6142对D1R的选择性得到了D1R基因敲除小鼠中无效应以及在D1R拮抗剂SCH-23390存在时反应被阻断的支持。此外,PF-6142改善了NMDA受体拮抗剂诱导的认知功能障碍的啮齿动物模型中的表现,如MK-801破坏的配对脉冲易化以及氯胺酮破坏的放射状臂迷宫中的工作记忆表现。同样,PF-6142逆转了氯胺酮诱导的NHP在执行空间延迟识别任务中的缺陷。重要的是,在预测啮齿动物抗精神病样效应的试验中,包括前脉冲抑制和条件性回避反应,PF-6142并未改变利培酮的疗效。这些数据支持继续研发基于非儿茶酚胺的D1R激动剂,用于治疗与包括精神分裂症在内的脑部疾病相关的认知障碍。