Ashford Roland T, Muchowski Jakub, Koylass Mark, Scholz Holger C, Whatmore Adrian M
Department of Bacteriology, Animal and Plant Health Agency, Weybridge, United Kingdom.
Department of Bacteriology and Toxinology, Bundeswehr Institute of Microbiology, Munich, Germany.
Front Microbiol. 2020 Jul 14;11:1329. doi: 10.3389/fmicb.2020.01329. eCollection 2020.
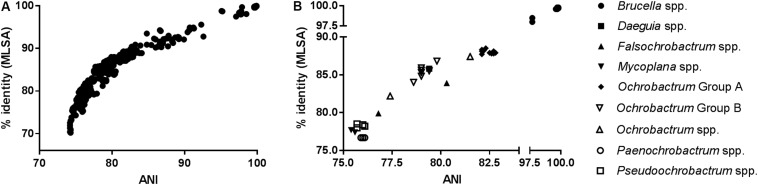
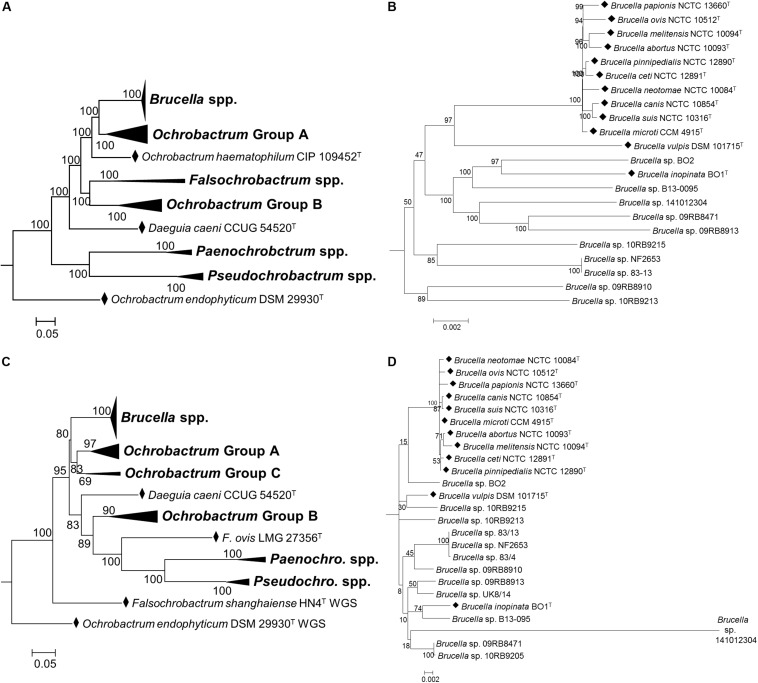
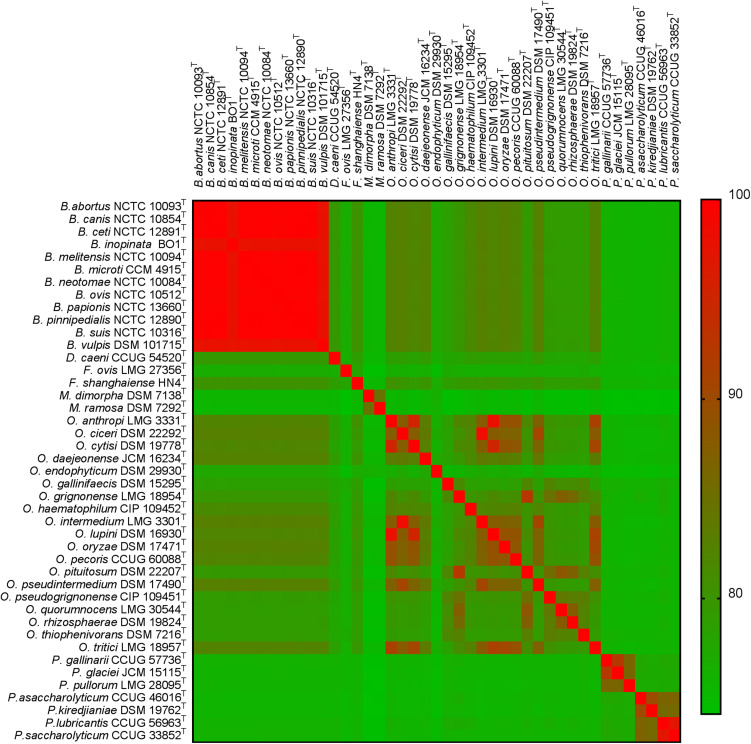
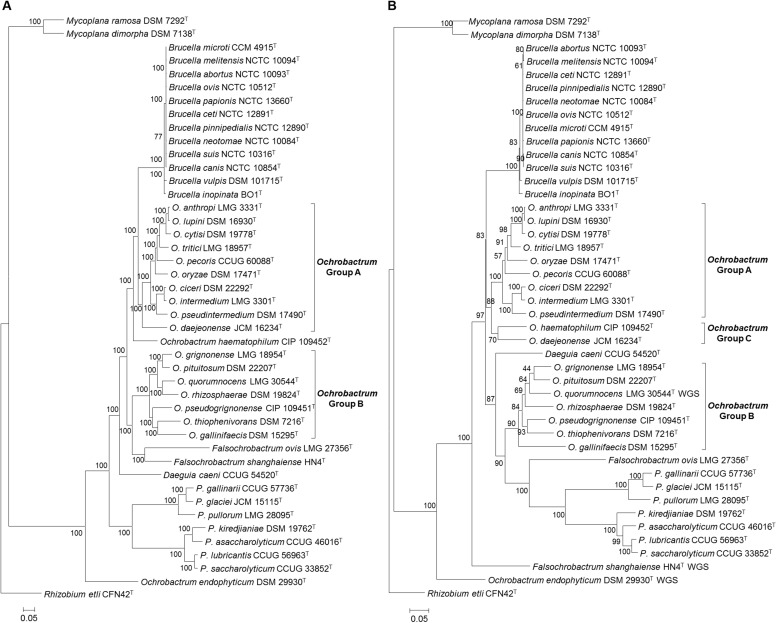
The bacterial family is currently composed of seven genera, including species of the genus , a number of which are significant veterinary and zoonotic pathogens. The bacteriological identification of pathogenic spp. may be hindered by their close phenotypic similarity to other members of the , particularly of the genus . Additionally, a number of novel atypical taxa have recently been identified, which exhibit greater genetic diversity than observed within the previously described species, and which share genomic features with organisms outside of the genus. Furthermore, previous work has indicated that the genus is polyphyletic, raising further questions regarding the relationship between the genus and wider . We have applied whole genome sequencing (WGS) and pan-family multi-locus sequence analysis (MLSA) approaches to a comprehensive panel of type strains, in order to characterize relationships within the family. Phylogenies based on WGS core genome alignments were able to resolve phylogenetic relationships of 31 non- spp. type strains from within the family, alongside type strains of twelve species. A phylogeny based on concatenated pan-family MLSA data was largely consistent with WGS based analyses. Notably, recently described atypical isolates were consistently placed in a single clade with existing species, clearly distinct from all members of the genus and wider family. Both WGS and MLSA methods closely grouped spp. with a sub-set of species. However, results also confirmed that the genus is polyphyletic, with seven species forming a separate grouping. The pan-family MLSA scheme was subsequently applied to a panel of 50 field strains of the family , isolated from a wide variety of sources. This analysis confirmed the utility of the pan- MLSA scheme in placing field isolates in relation to recognized type strains. However, a significant number of these isolates did not cluster with currently identified type strains, suggesting the existence of additional taxonomic diversity within some members of the . The WGS and pan-family MLSA approaches applied here provide valuable tools for resolving the identity and phylogenetic relationships of isolates from an expanding bacterial family containing a number of important pathogens.
该细菌家族目前由七个属组成,包括 属的一些物种,其中许多是重要的兽医和人畜共患病原体。致病性 spp. 的细菌学鉴定可能会受到其与 属其他成员,特别是 属成员在表型上的相似性的阻碍。此外,最近鉴定出了一些新的非典型 分类群,它们表现出比先前描述的物种更大的遗传多样性,并且与该属以外的生物体共享基因组特征。此外,先前的研究表明 属是多系的,这就 属与更广泛的 之间的关系提出了进一步的问题。我们将全基因组测序(WGS)和泛家族多位点序列分析(MLSA)方法应用于一组全面的 型菌株,以表征该家族内的关系。基于WGS核心基因组比对的系统发育能够解析该家族内31个非 spp. 型菌株以及12个 物种的型菌株之间的系统发育关系。基于串联的泛家族MLSA数据的系统发育在很大程度上与基于WGS的分析一致。值得注意的是,最近描述的非典型 分离株始终与现有物种一起置于单个进化枝中,明显不同于 属和更广泛家族的所有成员。WGS和MLSA方法都将 spp. 与一部分 物种紧密分组。然而,结果也证实 属是多系的,七个物种形成一个单独的分组。随后将泛家族MLSA方案应用于从各种来源分离的一组50个该家族的田间菌株。该分析证实了泛 MLSA方案在将田间分离株与公认的型菌株相关联方面的实用性。然而,这些分离株中有相当数量没有与目前鉴定的型菌株聚类,这表明在 属的一些成员中存在额外的分类多样性。这里应用的WGS和泛家族MLSA方法为解决来自一个包含许多重要病原体的不断扩大的细菌家族的分离株的身份和系统发育关系提供了有价值的工具。