Nordic Seed A/S, Grindsnabevej 25, 8300, Odder, Denmark.
Department of Agroecology, Faculty of Science and Technology, Aarhus University, Forsøgsvej 1, Flakkebjerg, 4200, Slagelse, Denmark.
Sci Rep. 2020 Aug 10;10(1):13475. doi: 10.1038/s41598-020-70406-2.
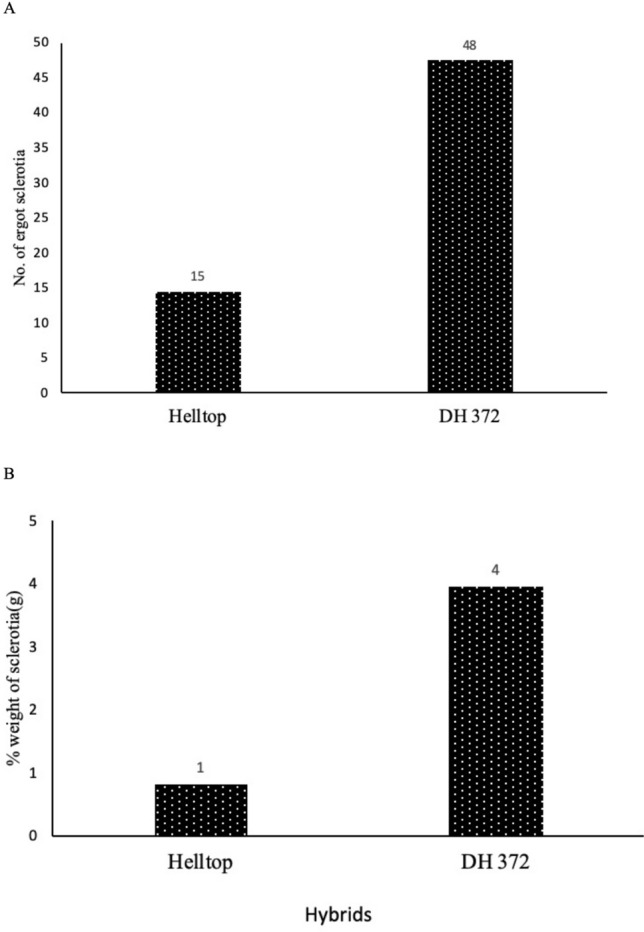
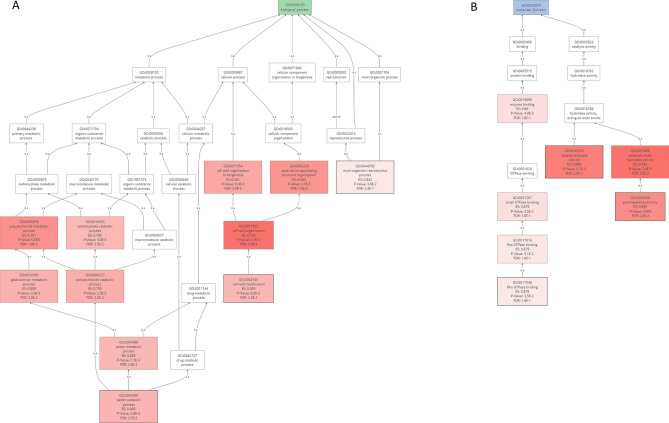
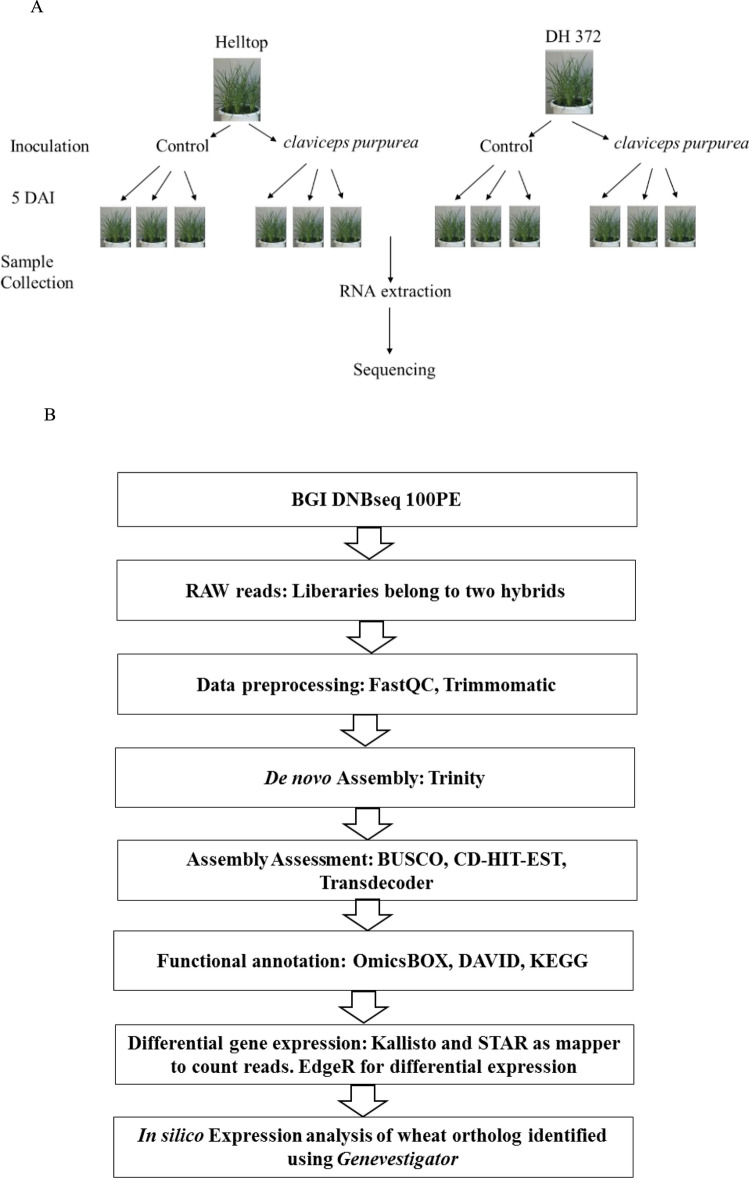
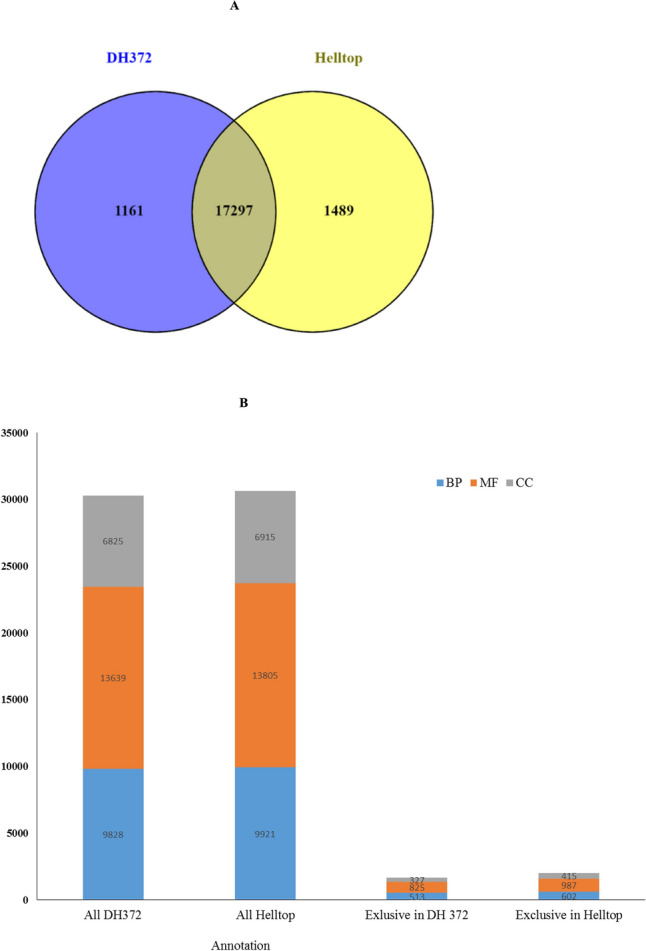
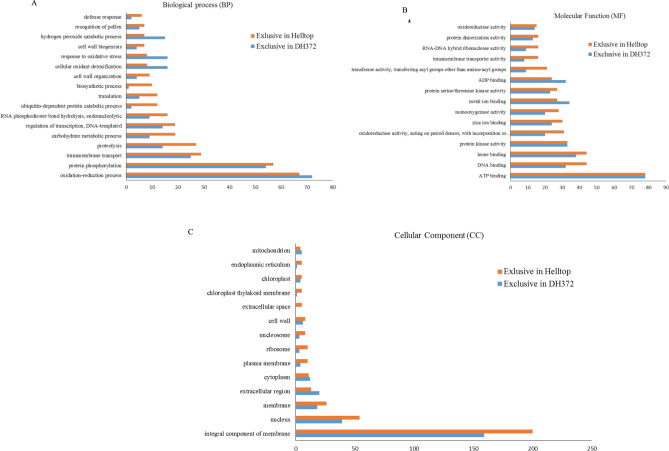
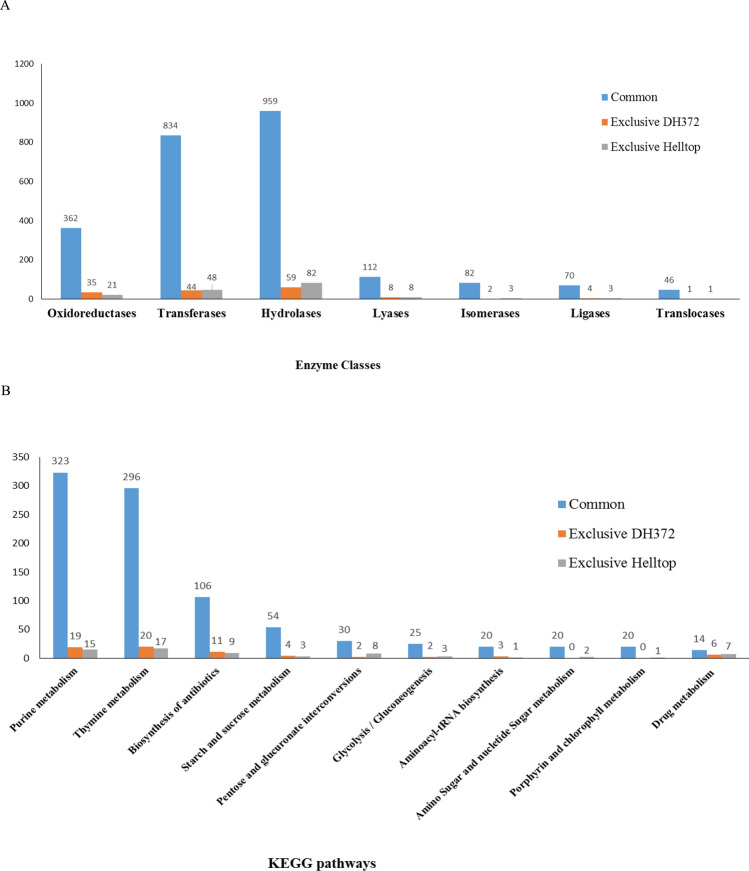
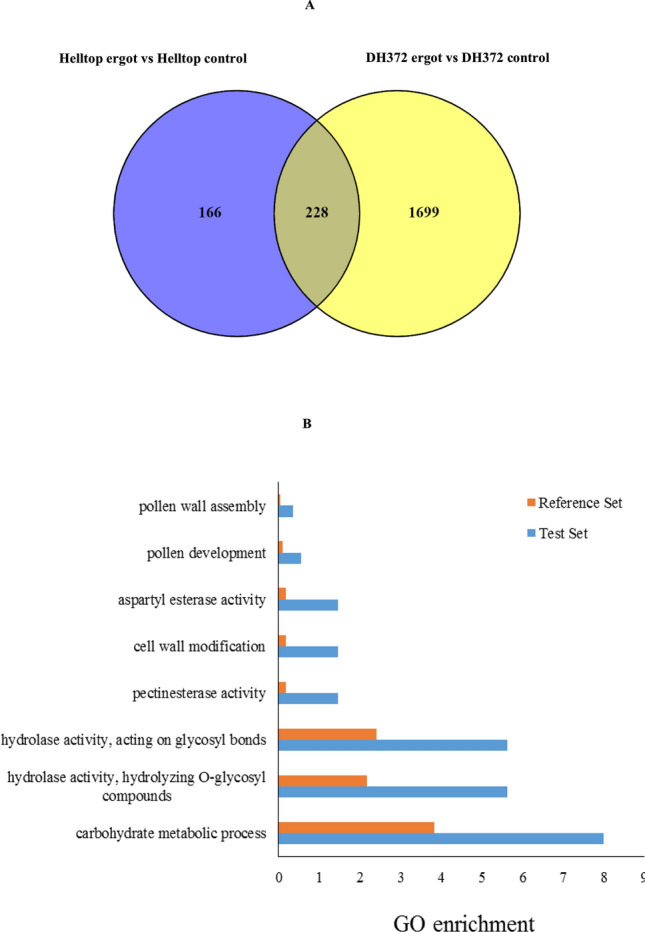
Rye is used as food, feed, and for bioenergy production and remain an essential grain crop for cool temperate zones in marginal soils. Ergot is known to cause severe problems in cross-pollinated rye by contamination of harvested grains. The molecular response of the underlying mechanisms of this disease is still poorly understood due to the complex infection pattern. RNA sequencing can provide astonishing details about the transcriptional landscape, hence we employed a transcriptomic approach to identify genes in the underlying mechanism of ergot infection in rye. In this study, we generated de novo assemblies from twelve biological samples of two rye hybrids with identified contrasting phenotypic responses to ergot infection. The final transcriptome of ergot susceptible (DH372) and moderately ergot resistant (Helltop) hybrids contain 208,690 and 192,116 contigs, respectively. By applying the BUSCO pipeline, we confirmed that these transcriptome assemblies contain more than 90% of gene representation of the available orthologue groups at Virdiplantae odb10. We employed a de novo assembled and the draft reference genome of rye to count the differentially expressed genes (DEGs) between the two hybrids with and without inoculation. The gene expression comparisons revealed that 228 genes were linked to ergot infection in both hybrids. The genome ontology enrichment analysis of DEGs associated them with metabolic processes, hydrolase activity, pectinesterase activity, cell wall modification, pollen development and pollen wall assembly. In addition, gene set enrichment analysis of DEGs linked them to cell wall modification and pectinesterase activity. These results suggest that a combination of different pathways, particularly cell wall modification and pectinesterase activity contribute to the underlying mechanism that might lead to resistance against ergot in rye. Our results may pave the way to select genetic material to improve resistance against ergot through better understanding of the mechanism of ergot infection at molecular level. Furthermore, the sequence data and de novo assemblies are valuable as scientific resources for future studies in rye.
黑麦被用作食物、饲料和生物能源生产,并且仍然是边缘土壤中凉爽温带地区的重要谷物作物。已知麦角菌会通过污染收获的谷物,在异花授粉的黑麦中引起严重问题。由于感染模式复杂,这种疾病的潜在机制的分子反应仍然知之甚少。RNA 测序可以提供关于转录景观的惊人细节,因此我们采用转录组学方法来鉴定黑麦麦角菌感染潜在机制中的基因。在这项研究中,我们从两个黑麦杂种的 12 个生物学样本中生成了从头组装,这些样本的表型对麦角菌感染有明显的不同反应。易受麦角菌感染的(DH372)和中度抗麦角菌的(Helltop)杂种的最终转录组分别包含 208690 和 192116 个连。通过应用 BUSCO 管道,我们证实这些转录组组装包含超过可用同源物组的 90%的代表在 Virdiplantae odb10 中。我们使用从头组装的和黑麦的草案参考基因组来计算有和没有接种的两个杂种之间的差异表达基因(DEGs)。基因表达比较表明,这两个杂种中与麦角菌感染相关的 228 个基因。与 DEGs 相关的基因本体富集分析将它们与代谢过程、水解酶活性、果胶酯酶活性、细胞壁修饰、花粉发育和花粉壁组装联系起来。此外,DEGs 相关的基因集富集分析将它们与细胞壁修饰和果胶酯酶活性联系起来。这些结果表明,不同途径的组合,特别是细胞壁修饰和果胶酯酶活性,有助于抵抗黑麦麦角菌的潜在机制。我们的研究结果可能为通过更好地了解麦角菌感染的分子机制来选择遗传物质以提高对麦角菌的抗性铺平道路。此外,序列数据和从头组装是未来黑麦研究的有价值的科学资源。