Center for Computational and Integrative Biology, Rutgers University, Camden, NJ, USA.
Department of Pharmacology and Toxicology, Ernest Mario School of Pharmacy, Rutgers University, Piscataway, NJ, USA.
Lab Invest. 2021 Apr;101(4):490-502. doi: 10.1038/s41374-020-00477-2. Epub 2020 Aug 10.
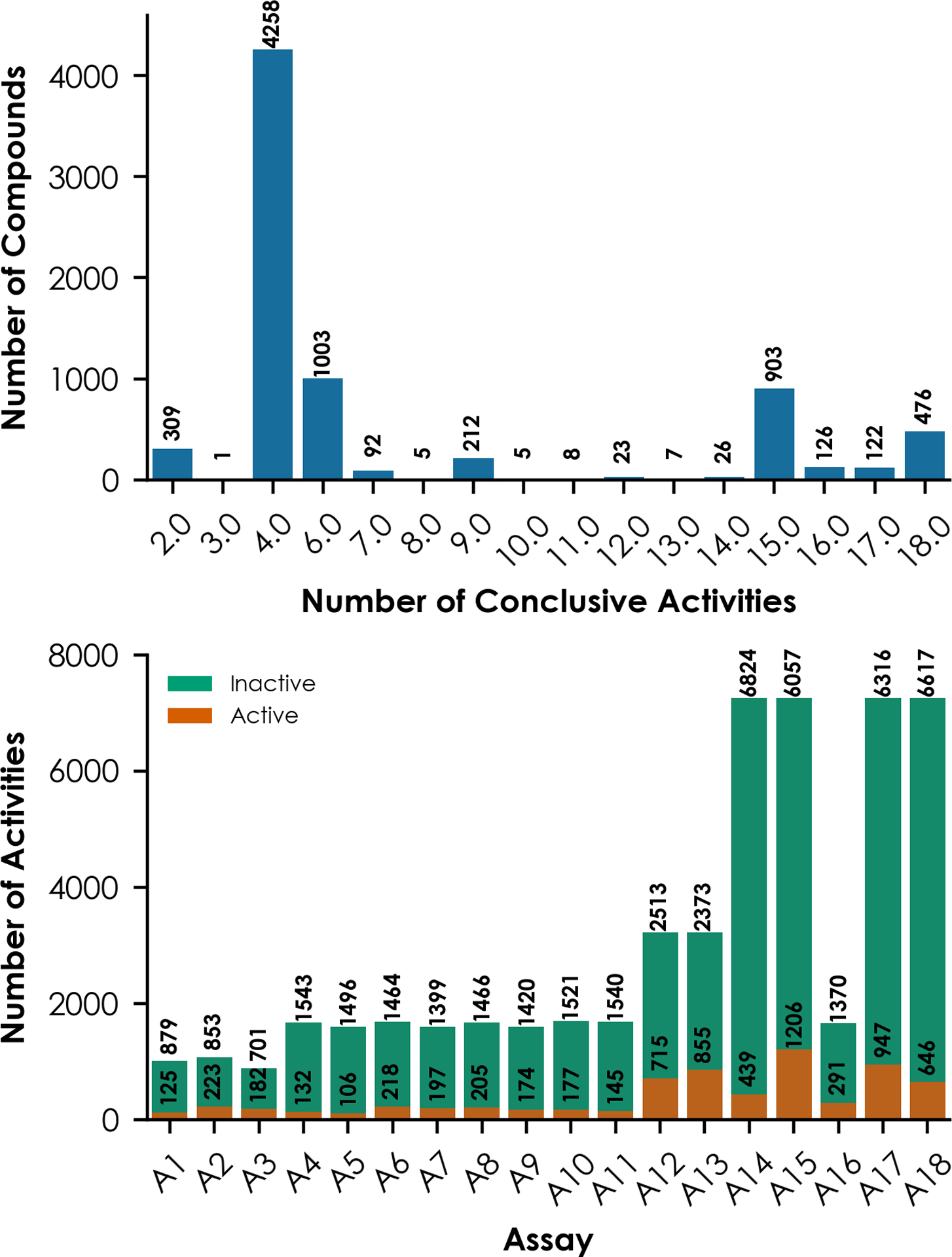
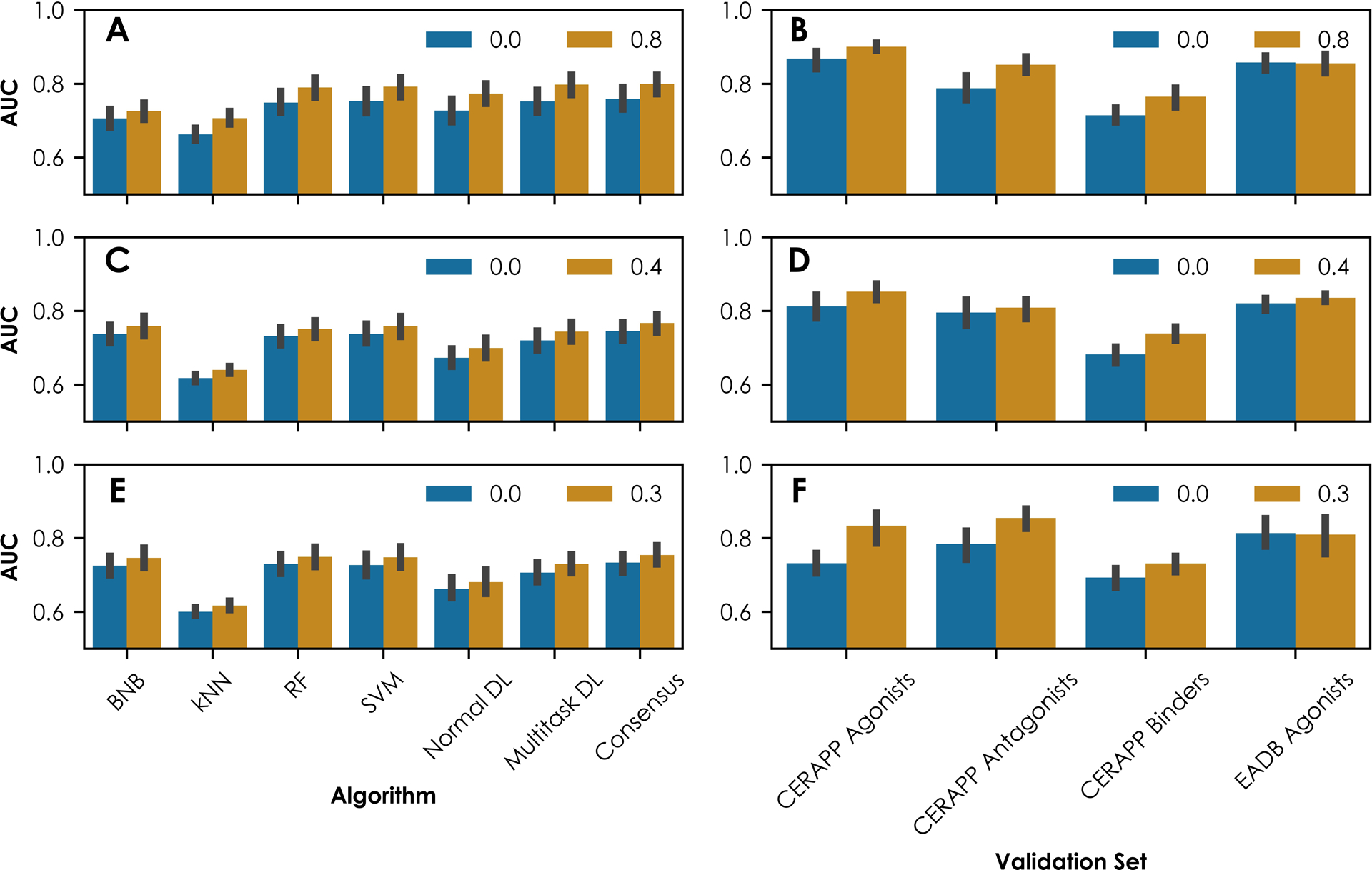
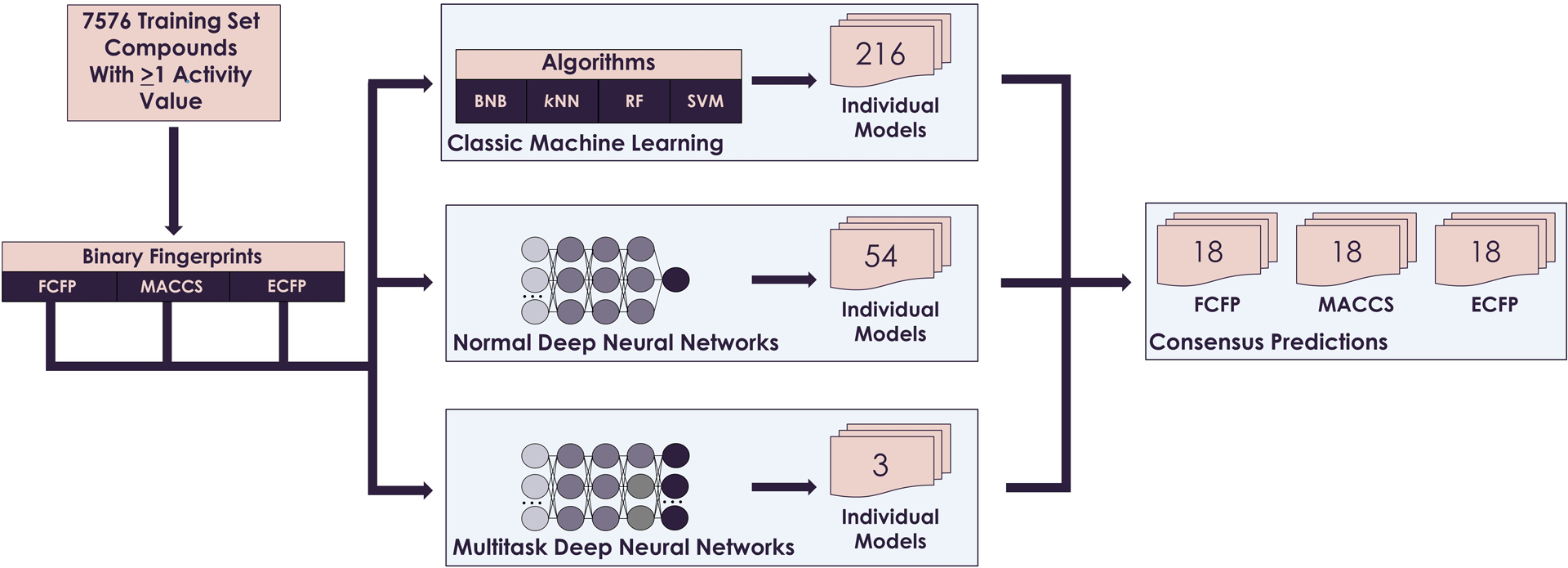
As defined by the World Health Organization, an endocrine disruptor is an exogenous substance or mixture that alters function(s) of the endocrine system and consequently causes adverse health effects in an intact organism, its progeny, or (sub)populations. Traditional experimental testing regimens to identify toxicants that induce endocrine disruption can be expensive and time-consuming. Computational modeling has emerged as a promising and cost-effective alternative method for screening and prioritizing potentially endocrine-active compounds. The efficient identification of suitable chemical descriptors and machine-learning algorithms, including deep learning, is a considerable challenge for computational toxicology studies. Here, we sought to apply classic machine-learning algorithms and deep-learning approaches to a panel of over 7500 compounds tested against 18 Toxicity Forecaster assays related to nuclear estrogen receptor (ERα and ERβ) activity. Three binary fingerprints (Extended Connectivity FingerPrints, Functional Connectivity FingerPrints, and Molecular ACCess System) were used as chemical descriptors in this study. Each descriptor was combined with four machine-learning and two deep- learning (normal and multitask neural networks) approaches to construct models for all 18 ER assays. The resulting model performance was evaluated using the area under the receiver- operating curve (AUC) values obtained from a fivefold cross-validation procedure. The results showed that individual models have AUC values that range from 0.56 to 0.86. External validation was conducted using two additional sets of compounds (n = 592 and n = 966) with established interactions with nuclear ER demonstrated through experimentation. An agonist, antagonist, or binding score was determined for each compound by averaging its predicted probabilities in relevant assay models as an external validation, yielding AUC values ranging from 0.63 to 0.91. The results suggest that multitask neural networks offer advantages when modeling mechanistically related endpoints. Consensus predictions based on the average values of individual models remain the best modeling strategy for computational toxicity evaluations.
世界卫生组织将内分泌干扰物定义为一种外源性物质或混合物,它改变了内分泌系统的功能,从而对完整的生物体、其后代或(亚)种群造成不良的健康影响。传统的实验测试方案用于识别诱导内分泌干扰的有毒物质可能既昂贵又耗时。计算建模已成为筛选和优先考虑潜在具有内分泌活性的化合物的一种有前途且具有成本效益的替代方法。对于计算毒理学研究来说,有效地识别合适的化学描述符和机器学习算法,包括深度学习,是一个相当大的挑战。在这里,我们试图将经典机器学习算法和深度学习方法应用于超过 7500 种化合物的面板,这些化合物针对与核雌激素受体(ERα 和 ERβ)活性相关的 18 种毒性预测器测定进行了测试。在这项研究中,使用了三种二进制指纹(扩展连接指纹、功能连接指纹和分子访问系统)作为化学描述符。每个描述符都与四种机器学习和两种深度学习(普通和多任务神经网络)方法相结合,为所有 18 个 ER 测定构建模型。使用来自五重交叉验证过程的接收者操作特征曲线 (AUC) 值评估所得模型的性能。结果表明,单个模型的 AUC 值范围从 0.56 到 0.86。使用通过实验证明与核 ER 具有相互作用的另外两组化合物(n=592 和 n=966)进行了外部验证。通过在相关测定模型中平均其预测概率来确定每个化合物的激动剂、拮抗剂或结合评分作为外部验证,AUC 值范围从 0.63 到 0.91。结果表明,多任务神经网络在模拟具有机制相关性的终点时具有优势。基于个体模型平均值的共识预测仍然是计算毒性评估的最佳建模策略。