Department of Neurology, University of Michigan, 109 Zina Pitcher Pl., Ann Arbor, MI, 48109, USA.
Department of Computational Medicine & Bioinformatics, University of Michigan, Ann Arbor, MI, 48109, USA.
Cerebellum. 2021 Feb;20(1):41-53. doi: 10.1007/s12311-020-01179-7.
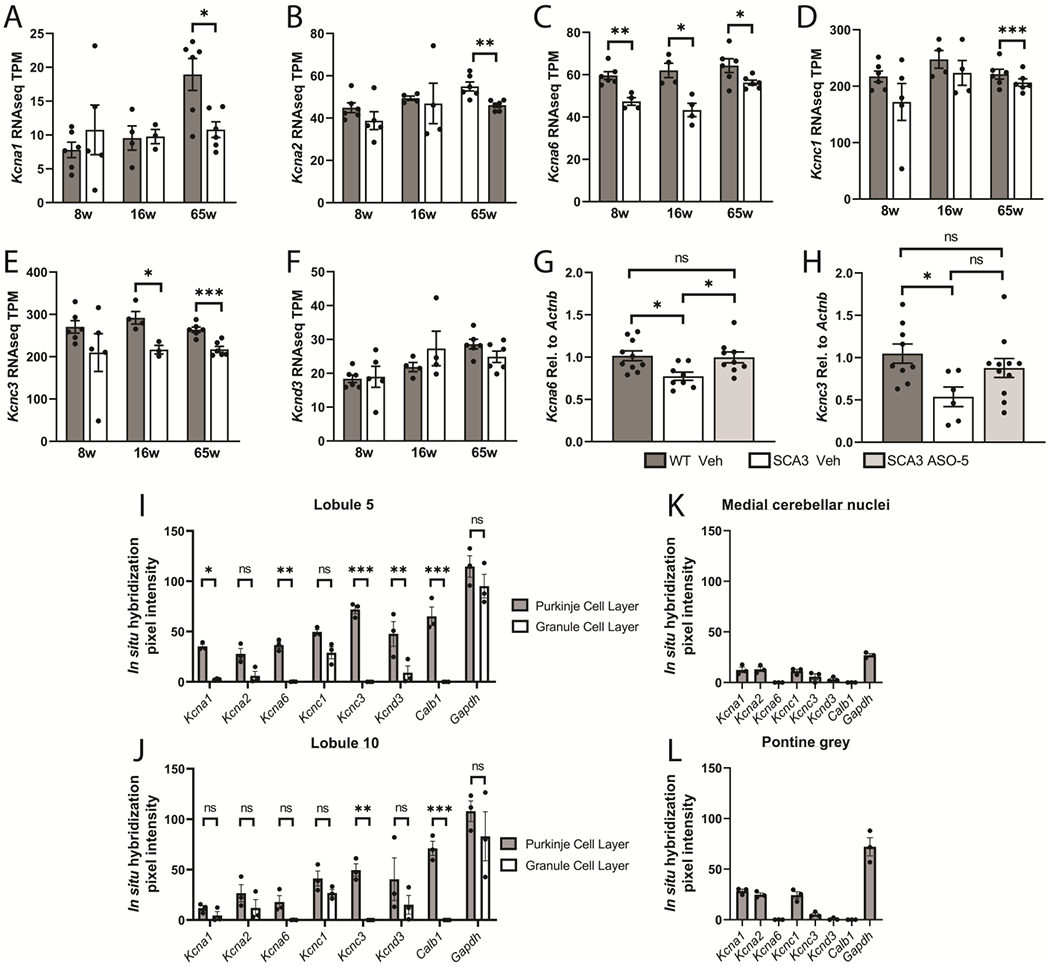
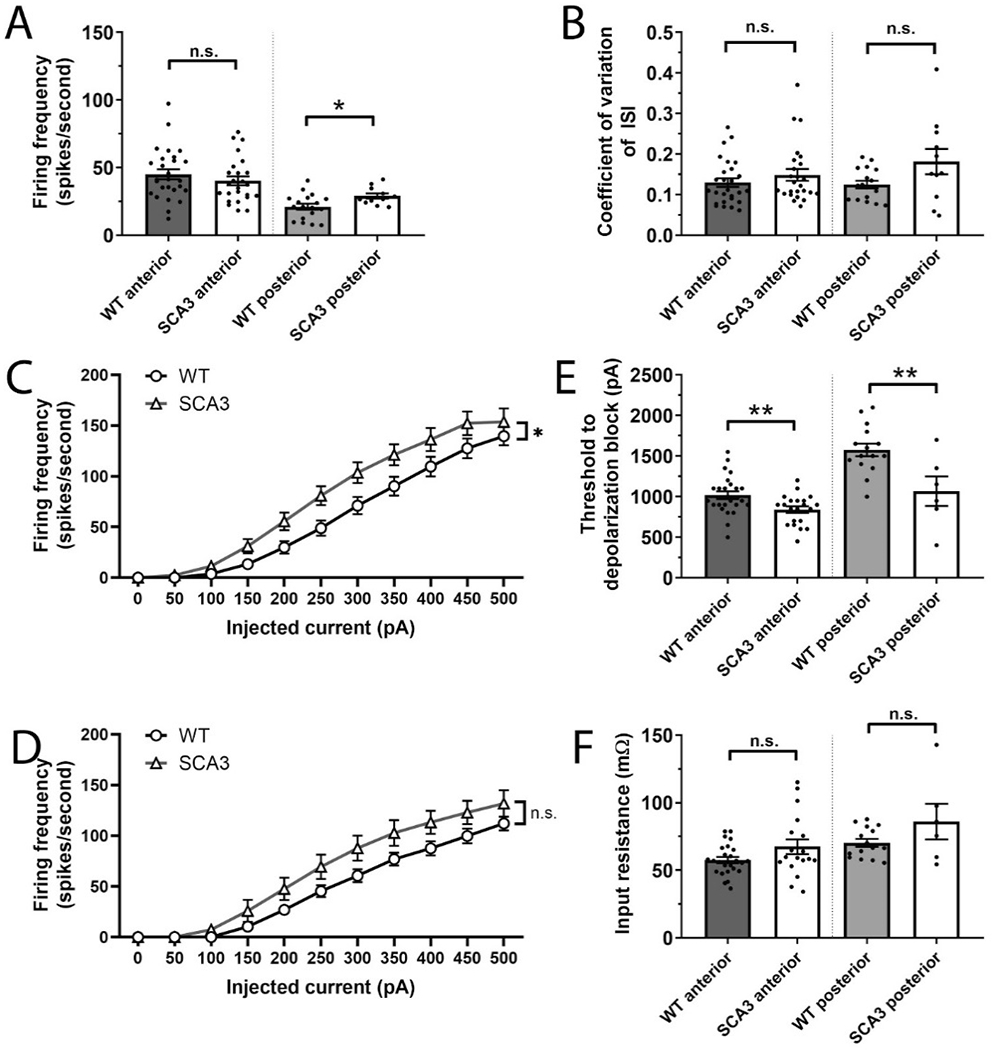
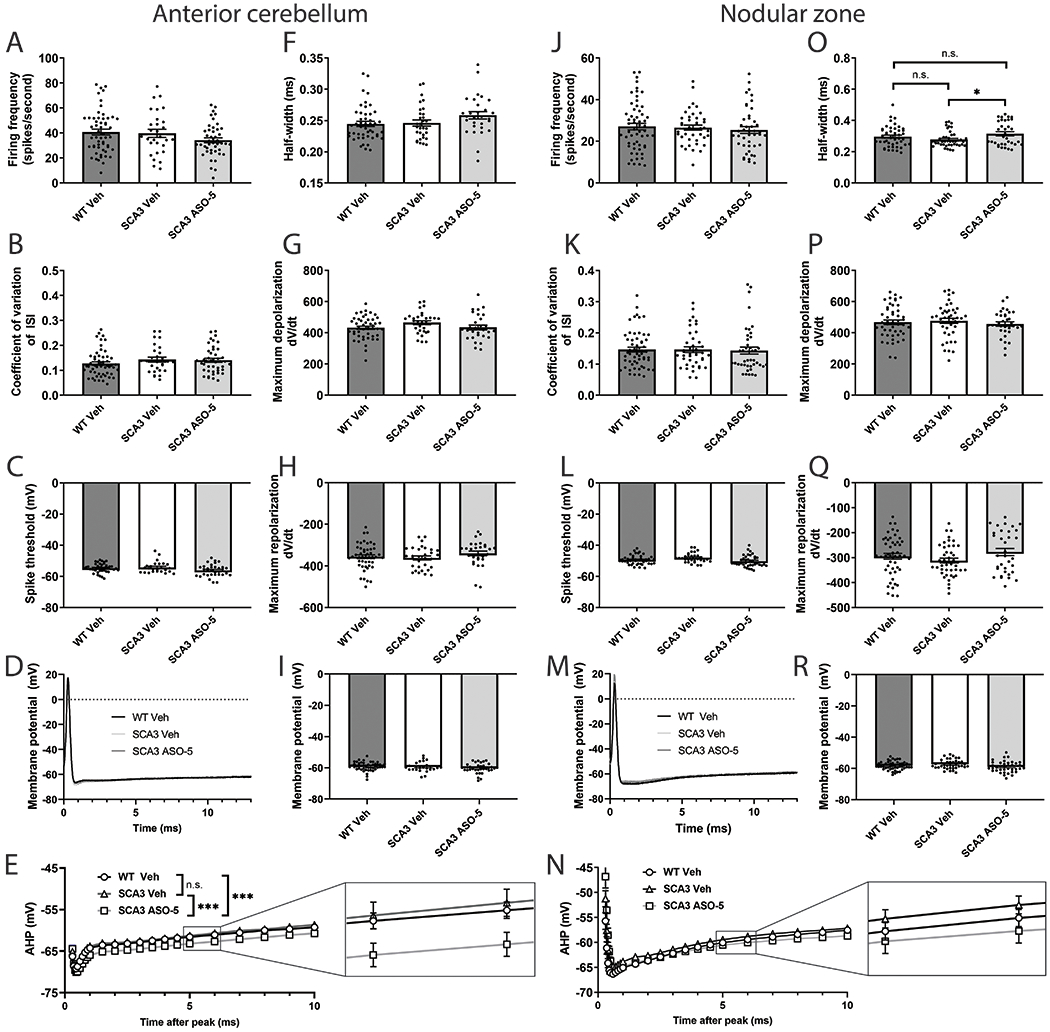
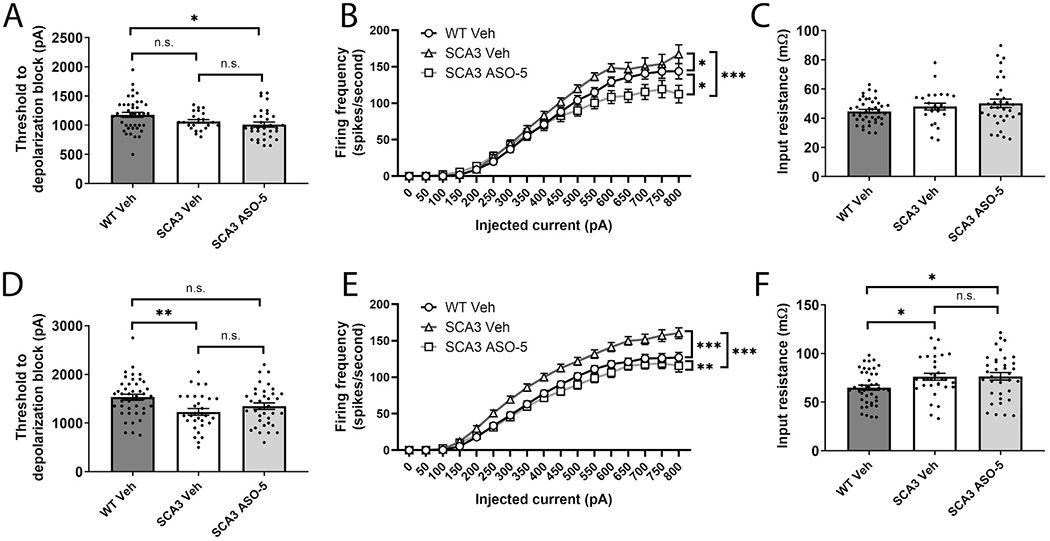
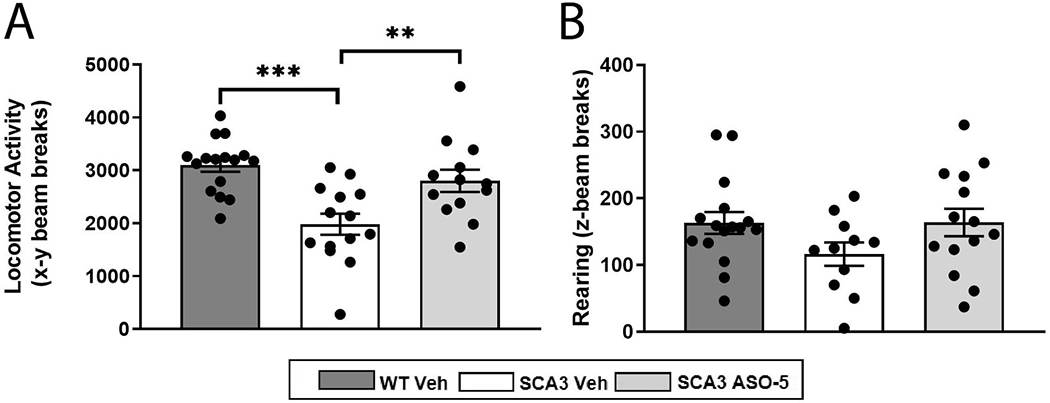
Spinocerebellar ataxia type 3 (SCA3) is the second-most common CAG repeat disease, caused by a glutamine-encoding expansion in the ATXN3 protein. SCA3 is characterized by spinocerebellar degeneration leading to progressive motor incoordination and early death. Previous studies suggest that potassium channel dysfunction underlies early abnormalities in cerebellar cortical Purkinje neuron firing in SCA3. However, cerebellar cortical degeneration is often modest both in the human disease and mouse models of SCA3, raising uncertainty about the role of cerebellar dysfunction in SCA3. Here, we address this question by investigating Purkinje neuron excitability in SCA3. In early-stage SCA3 mice, we confirm a previously identified increase in excitability of cerebellar Purkinje neurons and associate this excitability with reduced transcripts of two voltage-gated potassium (K) channels, Kcna6 and Kcnc3, as well as motor impairment. Intracerebroventricular delivery of antisense oligonucleotides (ASO) to reduce mutant ATXN3 restores normal excitability to SCA3 Purkinje neurons and rescues transcript levels of Kcna6 and Kcnc3. Interestingly, while an even broader range of K channel transcripts shows reduced levels in late-stage SCA3 mice, cerebellar Purkinje neuron physiology was not further altered despite continued worsening of motor impairment. These results suggest the progressive motor phenotype observed in SCA3 may not reflect ongoing changes in the cerebellar cortex but instead dysfunction of other neuronal structures within and beyond the cerebellum. Nevertheless, the early rescue of both K channel expression and neuronal excitability by ASO treatment suggests that cerebellar cortical dysfunction contributes meaningfully to motor dysfunction in SCA3.
脊髓小脑性共济失调 3 型(SCA3)是第二常见的 CAG 重复疾病,由 ATXN3 蛋白中谷氨酰胺编码扩展引起。SCA3 的特征是脊髓小脑变性,导致进行性运动协调障碍和早逝。先前的研究表明,钾通道功能障碍是 SCA3 小脑皮质浦肯野神经元放电早期异常的基础。然而,在人类疾病和 SCA3 小鼠模型中,小脑皮质变性通常都不明显,这使得小脑功能障碍在 SCA3 中的作用存在不确定性。在这里,我们通过研究 SCA3 中的浦肯野神经元兴奋性来解决这个问题。在 SCA3 早期阶段的小鼠中,我们确认了先前确定的小脑浦肯野神经元兴奋性增加,并将这种兴奋性与两种电压门控钾 (K) 通道 Kcna6 和 Kcnc3 的转录物减少以及运动障碍相关联。鞘内给予反义寡核苷酸 (ASO) 以减少突变型 ATXN3,可使 SCA3 浦肯野神经元恢复正常的兴奋性,并挽救 Kcna6 和 Kcnc3 的转录物水平。有趣的是,尽管在晚期 SCA3 小鼠中甚至更广泛的 K 通道转录物水平降低,但小脑浦肯野神经元生理学没有进一步改变,尽管运动障碍持续恶化。这些结果表明,在 SCA3 中观察到的进行性运动表型可能不反映小脑皮质的持续变化,而是小脑内部和超越小脑的其他神经元结构的功能障碍。然而,ASO 治疗早期对 K 通道表达和神经元兴奋性的挽救表明,小脑皮质功能障碍对 SCA3 中的运动功能障碍有重要意义。