Department of Informatics, Systems and Communication, University of Milano-Bicocca, Milan, Italy.
Department of Physiology and Biophysics, Tri-I Computational Biology & Medicine Graduate Program, Weill Cornell Medicine of Cornell University, New York, NY 10021, USA.
Bioinformatics. 2021 Apr 20;37(3):326-333. doi: 10.1093/bioinformatics/btaa722.
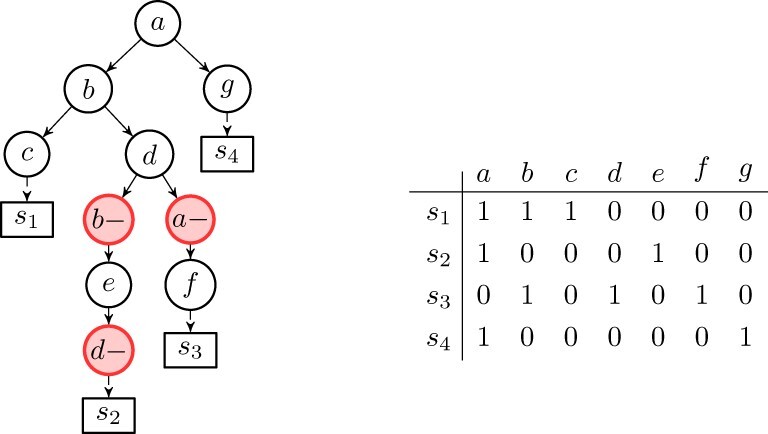
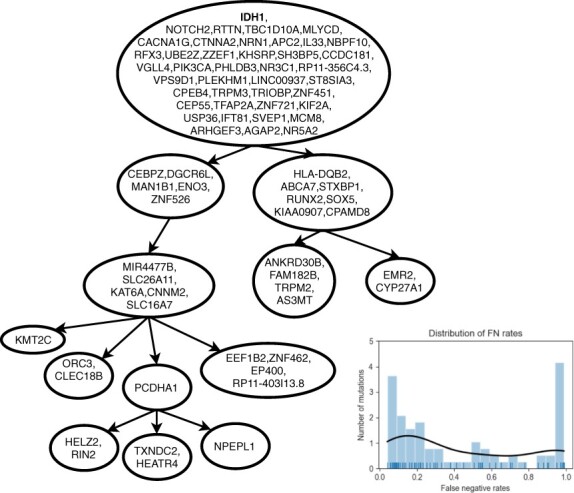
In recent years, the well-known Infinite Sites Assumption has been a fundamental feature of computational methods devised for reconstructing tumor phylogenies and inferring cancer progressions. However, recent studies leveraging single-cell sequencing (SCS) techniques have shown evidence of the widespread recurrence and, especially, loss of mutations in several tumor samples. While there exist established computational methods that infer phylogenies with mutation losses, there remain some advancements to be made.
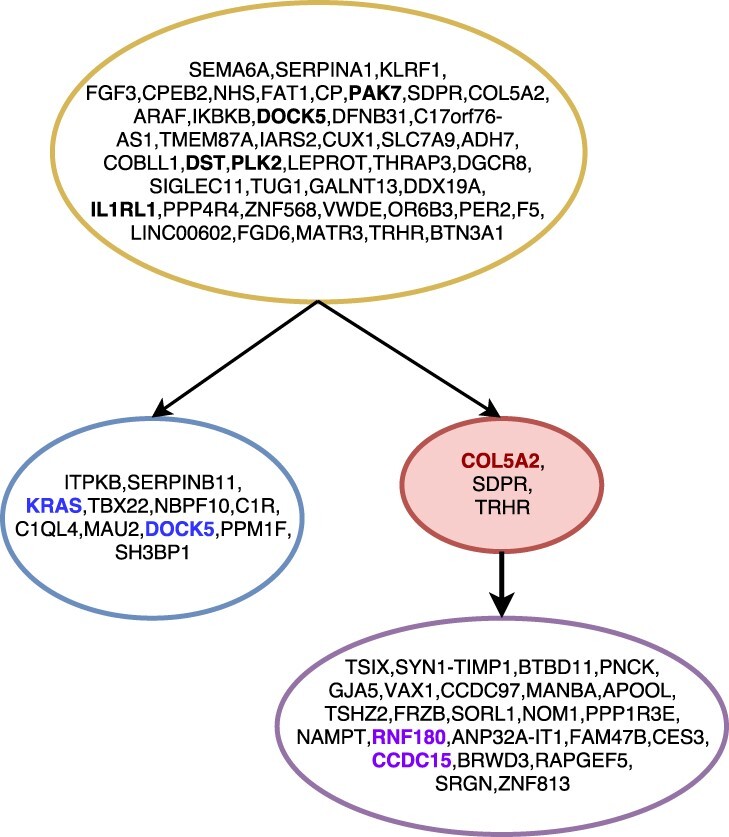
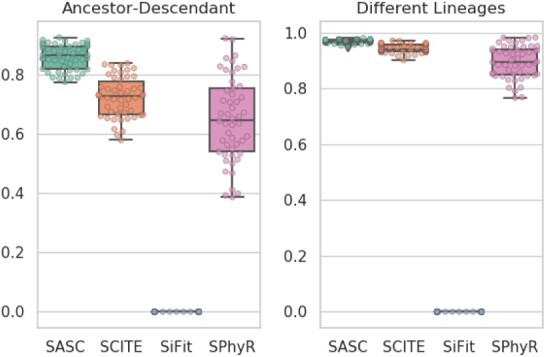
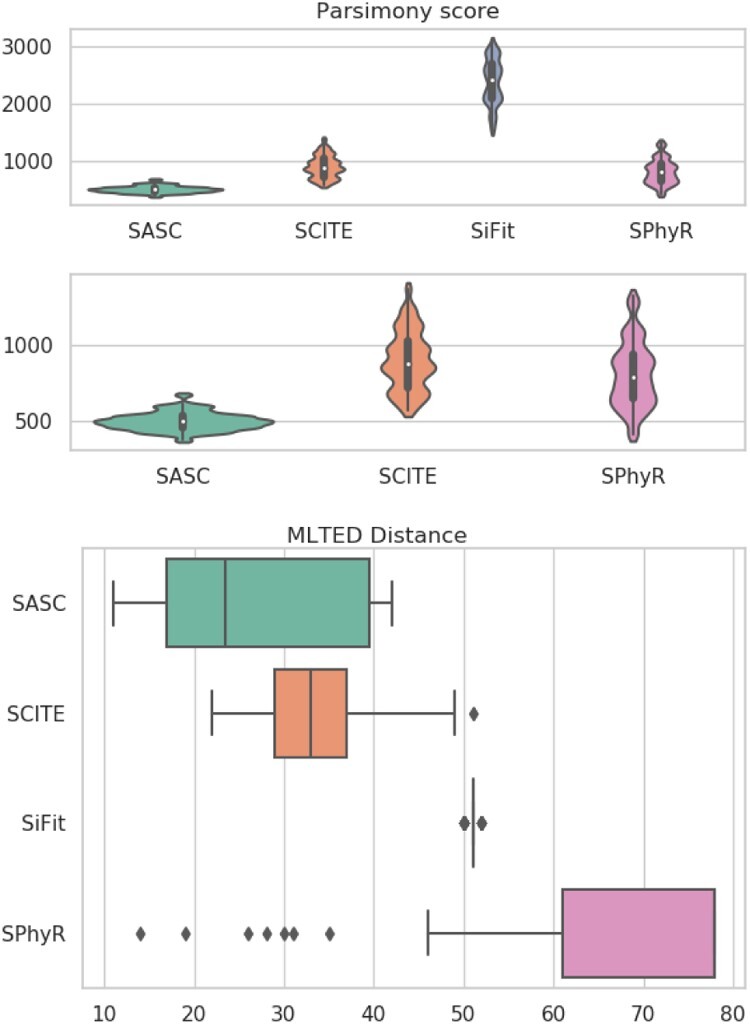
We present Simulated Annealing Single-Cell inference (SASC): a new and robust approach based on simulated annealing for the inference of cancer progression from SCS datasets. In particular, we introduce an extension of the model of evolution where mutations are only accumulated, by allowing also a limited amount of mutation loss in the evolutionary history of the tumor: the Dollo-k model. We demonstrate that SASC achieves high levels of accuracy when tested on both simulated and real datasets and in comparison with some other available methods.
The SASC tool is open source and available at https://github.com/sciccolella/sasc.
Supplementary data are available at Bioinformatics online.
近年来,著名的无限站点假设已成为为重建肿瘤系统发育和推断癌症进展而设计的计算方法的基本特征。然而,最近利用单细胞测序 (SCS) 技术的研究表明,在多个肿瘤样本中广泛存在突变的重现,特别是丢失。虽然存在推断具有突变丢失的系统发育的既定计算方法,但仍有一些改进需要进行。
我们提出了模拟退火单细胞推断 (SASC):一种新的稳健方法,基于模拟退火从 SCS 数据集推断癌症进展。特别是,我们通过允许肿瘤进化史中的突变丢失有限量,引入了突变仅累积的进化模型的扩展:多洛-克模型。我们证明,当在模拟和真实数据集上进行测试并与其他一些可用方法进行比较时,SASC 达到了很高的准确性水平。
SASC 工具是开源的,可在 https://github.com/sciccolella/sasc 上获得。
补充数据可在 Bioinformatics 在线获得。