Department of Biochemistry, University of Illinois at Urbana-Champaign, Urbana, 61801, IL, USA.
Fujian Key Laboratory for Translational Research in Cancer and Neurodegenerative Diseases, Institute for Translational Medicine, School of Basic Medical Sciences, Fujian Medical University, Fuzhou, 350122, China.
Cell Death Dis. 2020 Aug 21;11(8):667. doi: 10.1038/s41419-020-02894-z.
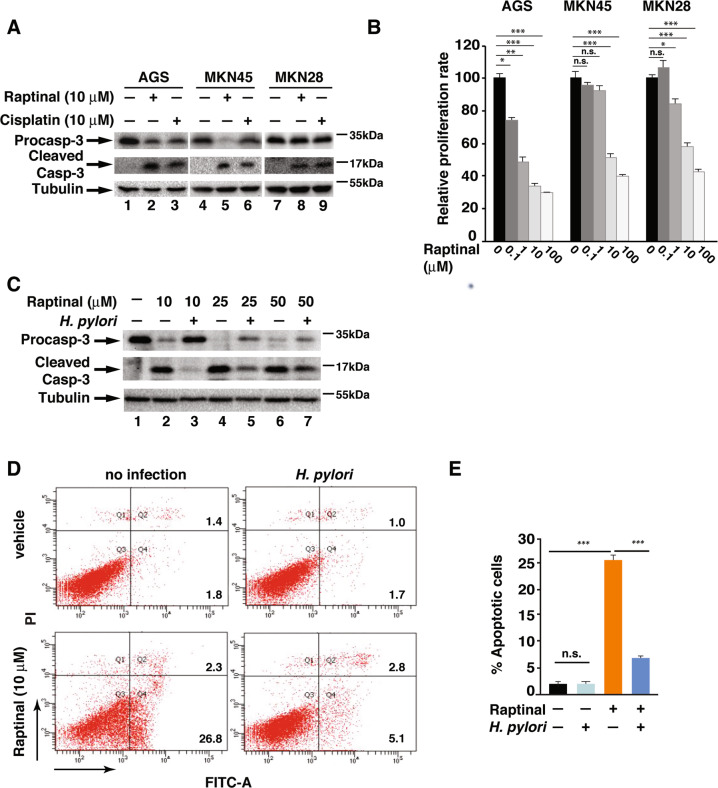
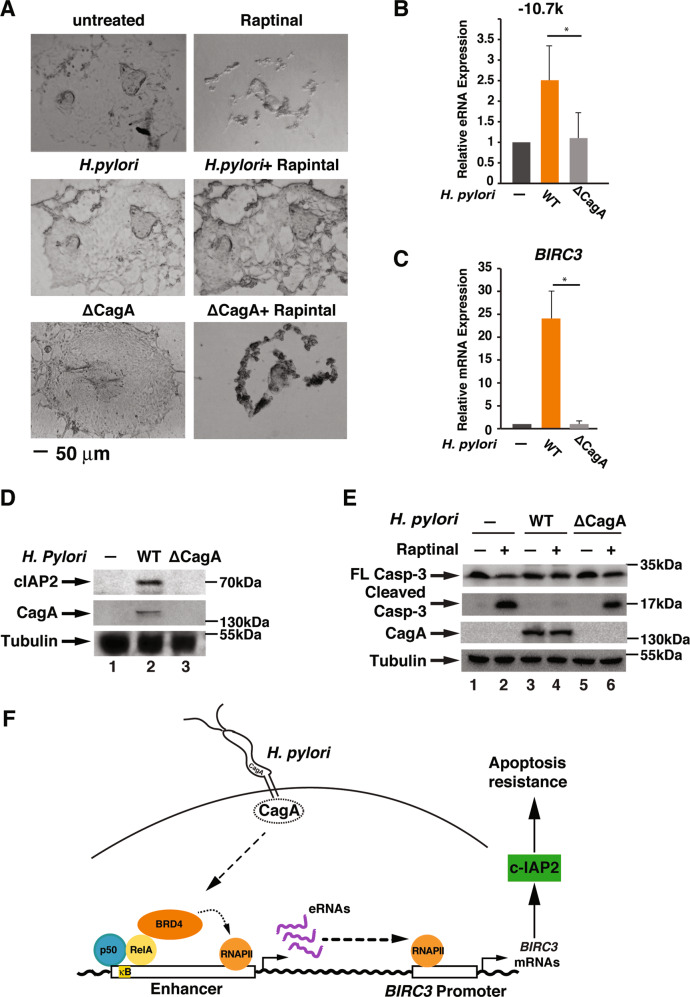
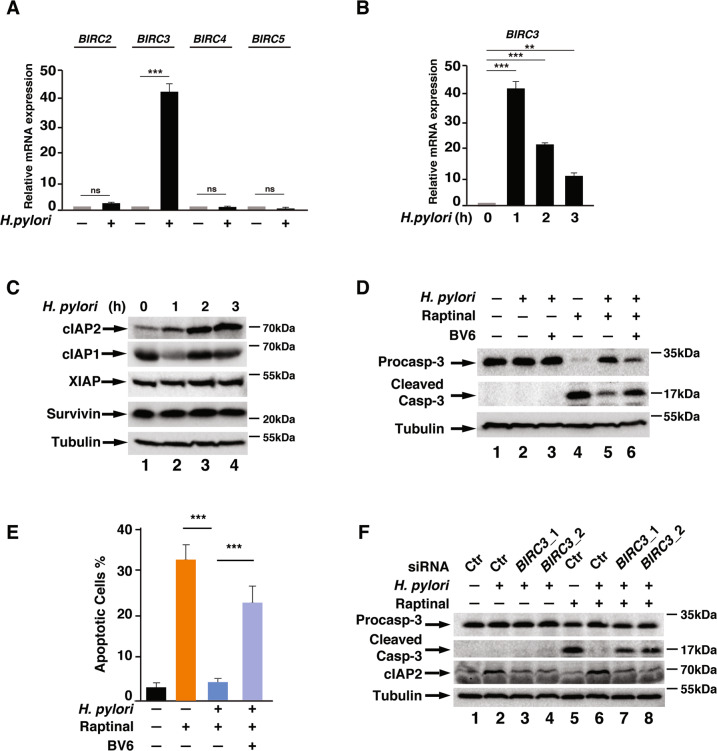
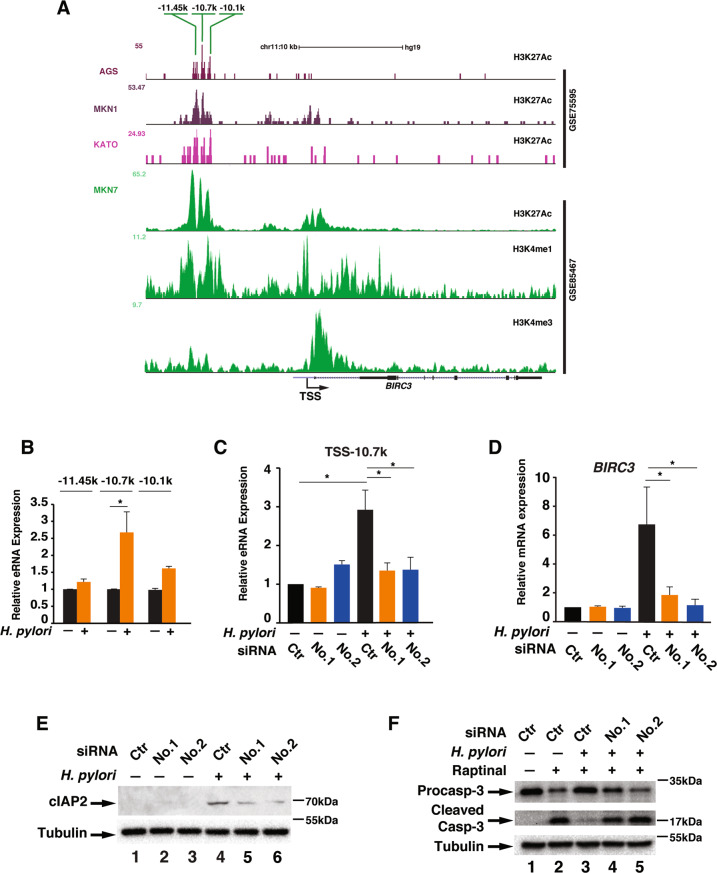
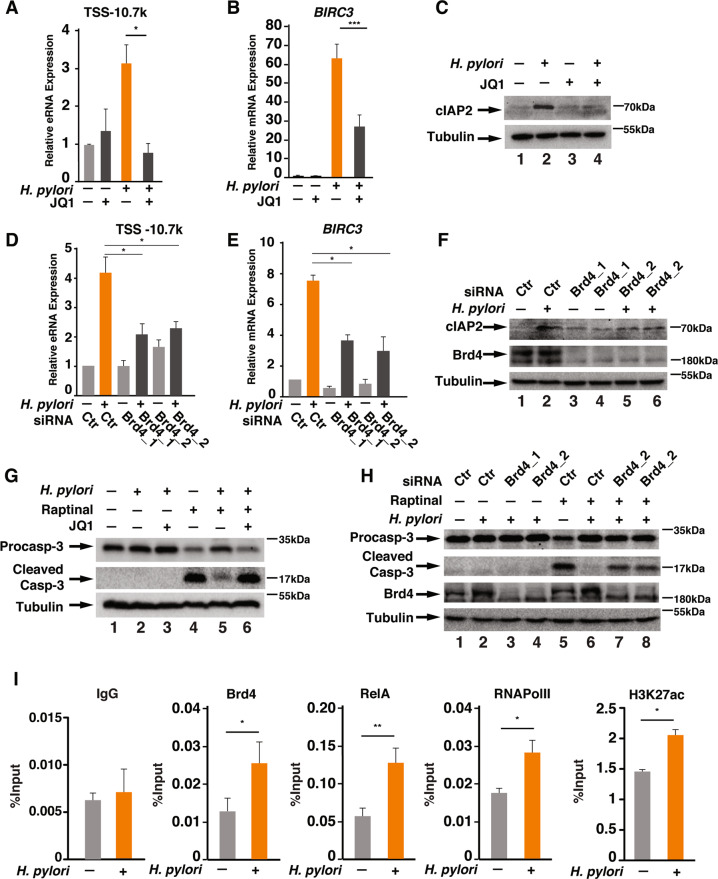
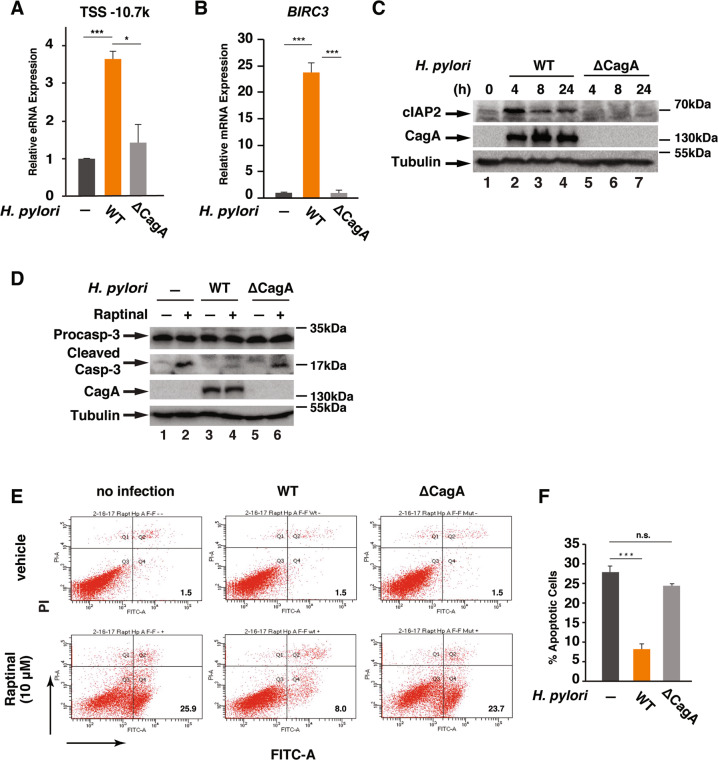
H. pylori infection is one of the leading causes of gastric cancer and the pathogenicity of H. pylori infection is associated with its ability to induce chronic inflammation and apoptosis resistance. While H. pylori infection-induced expression of pro-inflammatory cytokines for chronic inflammation is well studied, the molecular mechanism underlying the apoptosis resistance in infected cells is not well understood. In this study, we demonstrated that H. pylori infection-induced apoptosis resistance in gastric epithelial cells triggered by Raptinal, a drug that directly activates caspase-3. This resistance resulted from the induction of cIAP2 (encoded by BIRC3) since depletion of BIRC3 by siRNA or inhibition of cIAP2 via BV6 reversed H. pylori-suppressed caspase-3 activation. The induction of cIAP2 was regulated by H. pylori-induced BIRC3 eRNA synthesis. Depletion of BIRC3 eRNA decreased H. pylori-induced cIAP2 and reversed H. pylori-suppressed caspase-3 activation. Mechanistically, H. pylori stimulated the recruitment of bromodomain-containing factor Brd4 to the enhancer of BIRC3 and promoted BIRC3 eRNA and mRNA synthesis. Inhibition of Brd4 diminished the expression of BIRC3 eRNA and the anti-apoptotic response to H. pylori infection. Importantly, H. pylori isogenic cagA-deficient mutant failed to activate the synthesis of BIRC3 eRNA and the associated apoptosis resistance. Finally, in primary human gastric epithelial cells, H. pylori also induced resistance to Raptinal-triggered caspase-3 activation by activating the Brd4-dependent BIRC3 eRNA synthesis in a CagA-dependent manner. These results identify a novel function of Brd4 in H. pylori-mediated apoptosis resistance via activating BIRC3 eRNA synthesis, suggesting that Brd4 could be a potential therapeutic target for H. pylori-induced gastric cancer.
幽门螺杆菌感染是胃癌的主要病因之一,其致病性与诱导慢性炎症和抗细胞凋亡能力有关。虽然幽门螺杆菌感染诱导促炎细胞因子表达引起慢性炎症的机制已得到充分研究,但感染细胞中抗细胞凋亡的分子机制尚不清楚。在这项研究中,我们证明了 Raptinal(一种直接激活 caspase-3 的药物)触发的幽门螺杆菌感染诱导的胃上皮细胞抗凋亡作用。这种抵抗源自 cIAP2(由 BIRC3 编码)的诱导,因为 siRNA 耗尽 BIRC3 或通过 BV6 抑制 cIAP2 可逆转 H. pylori 抑制的 caspase-3 激活。cIAP2 的诱导受 H. pylori 诱导的 BIRC3 eRNA 合成调节。耗尽 BIRC3 eRNA 可降低 H. pylori 诱导的 cIAP2 并逆转 H. pylori 抑制的 caspase-3 激活。在机制上,幽门螺杆菌刺激含有溴结构域的因子 Brd4 募集到 BIRC3 的增强子,并促进 BIRC3 eRNA 和 mRNA 的合成。Brd4 的抑制减少了 BIRC3 eRNA 的表达和对 H. pylori 感染的抗凋亡反应。重要的是,cagA 缺陷型幽门螺杆菌突变体不能激活 BIRC3 eRNA 的合成和相关的抗凋亡作用。最后,在原代人胃上皮细胞中,幽门螺杆菌还通过以 CagA 依赖的方式激活 Brd4 依赖性 BIRC3 eRNA 合成,诱导对 Raptinal 触发的 caspase-3 激活的抗性。这些结果确定了 Brd4 在幽门螺杆菌介导的抗细胞凋亡中的新功能,通过激活 BIRC3 eRNA 合成,表明 Brd4 可能是治疗幽门螺杆菌诱导的胃癌的潜在靶点。