Feurtey Alice, Lorrain Cécile, Croll Daniel, Eschenbrenner Christoph, Freitag Michael, Habig Michael, Haueisen Janine, Möller Mareike, Schotanus Klaas, Stukenbrock Eva H
Environmental Genomics, Max Planck Institute for Evolutionary Biology, 24306, Plön, Germany.
Environmental Genomics, Christian-Albrechts University of Kiel, 24118, Kiel, Germany.
BMC Genomics. 2020 Aug 26;21(1):588. doi: 10.1186/s12864-020-06871-w.
Antagonistic co-evolution can drive rapid adaptation in pathogens and shape genome architecture. Comparative genome analyses of several fungal pathogens revealed highly variable genomes, for many species characterized by specific repeat-rich genome compartments with exceptionally high sequence variability. Dynamic genome structure may enable fast adaptation to host genetics. The wheat pathogen Zymoseptoria tritici with its highly variable genome, has emerged as a model organism to study genome evolution of plant pathogens. Here, we compared genomes of Z. tritici isolates and of sister species infecting wild grasses to address the evolution of genome composition and structure.
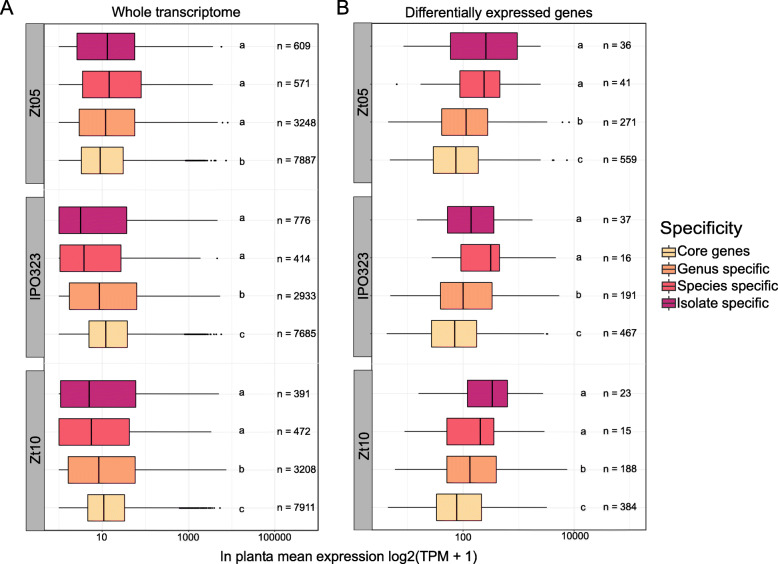
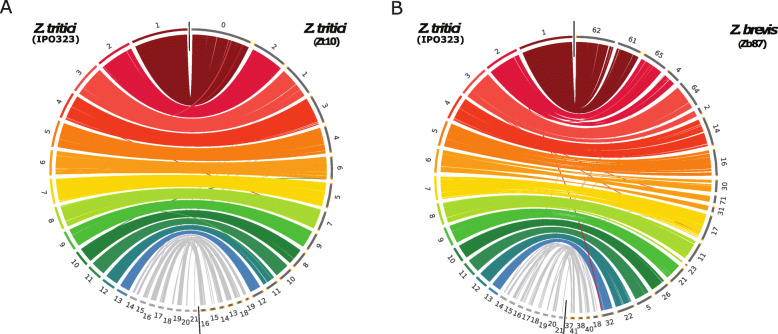
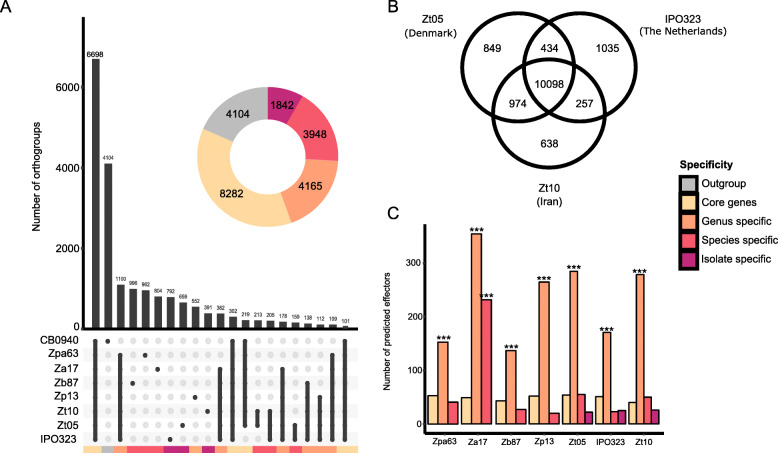
Using long-read technology, we sequenced and assembled genomes of Z. ardabiliae, Z. brevis, Z. pseudotritici and Z. passerinii, together with two isolates of Z. tritici. We report a high extent of genome collinearity among Zymoseptoria species and high conservation of genomic, transcriptomic and epigenomic signatures of compartmentalization. We identify high gene content variability both within and between species. In addition, such variability is mainly limited to the accessory chromosomes and accessory compartments. Despite strong host specificity and non-overlapping host-range between species, predicted effectors are mainly shared among Zymoseptoria species, yet exhibiting a high level of presence-absence polymorphism within Z. tritici. Using in planta transcriptomic data from Z. tritici, we suggest different roles for the shared orthologs and for the accessory genes during infection of their hosts.
Despite previous reports of high genomic plasticity in Z. tritici, we describe here a high level of conservation in genomic, epigenomic and transcriptomic composition and structure across the genus Zymoseptoria. The compartmentalized genome allows the maintenance of a functional core genome co-occurring with a highly variable accessory genome.
拮抗协同进化可推动病原体的快速适应并塑造基因组结构。对几种真菌病原体的比较基因组分析揭示了高度可变的基因组,许多物种的特征是具有特定的富含重复序列的基因组区域,其序列变异性极高。动态的基因组结构可能使病原体能够快速适应宿主遗传学。小麦病原体小麦叶锈菌(Zymoseptoria tritici)具有高度可变的基因组,已成为研究植物病原体基因组进化的模式生物。在此,我们比较了小麦叶锈菌分离株和感染野生禾本科植物的近缘物种的基因组,以探讨基因组组成和结构的进化。
利用长读长技术,我们对阿德比叶锈菌(Z. ardabiliae)、短叶锈菌(Z. brevis)、拟小麦叶锈菌(Z. pseudotritici)和雀麦叶锈菌(Z. passerinii)的基因组以及两个小麦叶锈菌分离株进行了测序和组装。我们报道了叶锈菌属物种间高度的基因组共线性以及分区的基因组、转录组和表观基因组特征的高度保守性。我们鉴定出物种内部和物种之间都存在高基因含量变异性。此外,这种变异性主要局限于副染色体和副基因组区域。尽管物种间具有很强的宿主特异性且宿主范围不重叠,但预测的效应子在叶锈菌属物种中主要是共享的,不过在小麦叶锈菌内部表现出高水平的存在/缺失多态性。利用小麦叶锈菌的植物体内转录组数据,我们提出了共享直系同源基因和副基因在感染宿主过程中的不同作用。
尽管之前有报道称小麦叶锈菌具有高基因组可塑性,但我们在此描述了叶锈菌属在基因组、表观基因组和转录组组成及结构方面的高度保守性。分区化的基因组允许维持一个功能核心基因组与一个高度可变的辅助基因组同时存在。