Lepuschitz Sarah, Weinmaier Thomas, Mrazek Katharina, Beisken Stephan, Weinberger Johannes, Posch Andreas E
Ares Genetics GmbH, Vienna, Austria.
Front Microbiol. 2020 Aug 5;11:1883. doi: 10.3389/fmicb.2020.01883. eCollection 2020.
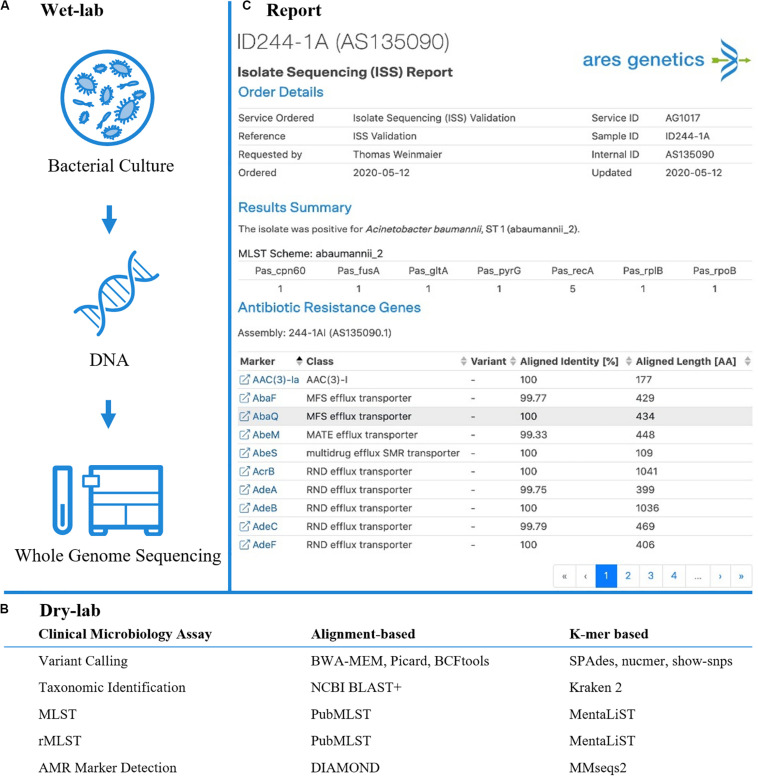
Next-generation sequencing (NGS) enables clinical microbiology assays such as molecular typing of bacterial isolates which is now routinely applied for infection control and epidemiology. Additionally, feasibility for NGS-based identification of antimicrobial resistance (AMR) markers as well as genetic prediction of antibiotic susceptibility testing results has been demonstrated. Various bioinformatics approaches enabling NGS-based clinical microbiology assays exist, but standardized, computationally efficient and scalable sample-to-results workflows including validated quality control parameters are still lacking. Bioinformatics analysis workflows based on k-mers have been shown to allow for fast and efficient analysis of large genomics data sets as obtained from microbial sequencing applications. We here demonstrate applicability of k-mer based clinical microbiology assays for whole-genome sequencing (WGS) including variant calling, taxonomic identification, bacterial typing as well as AMR marker detection. The wet-lab and dry-lab workflows were developed and validated in line with Clinical Laboratory Improvement Act (CLIA) guidelines for laboratory-developed tests (LDTs) on multi-drug resistant ESKAPE pathogens. The developed k-mer based workflow demonstrated ≥99.39% repeatability, ≥99.09% reproducibility and ≥99.76% accuracy for variant calling and applied assays as determined by intra-day and inter-day triplicate measurements. The limit of detection (LOD) across assays was found to be at 20× sequencing depth and 15× for AMR marker detection. Thorough benchmarking of the k-mer based workflow revealed analytical performance criteria are comparable to state-of-the-art alignment based workflows across clinical microbiology assays. Diagnostic sensitivity and specificity for multilocus sequence typing (MLST) and phylogenetic analysis were 100% for both approaches. For AMR marker detection, sensitivity and specificity were 95.29 and 99.78% for the k-mer based workflow as compared to 95.17 and 99.77% for the alignment-based approach. Summarizing, results illustrate that k-mer based analysis workflows enable a broad range of clinical microbiology assays, potentially not only for WGS-based typing and AMR gene detection but also genetic prediction of antibiotic susceptibility testing results.
下一代测序(NGS)推动了临床微生物学检测的发展,例如细菌分离株的分子分型,目前已常规应用于感染控制和流行病学研究。此外,基于NGS识别抗菌药物耐药性(AMR)标记以及对抗生素敏感性测试结果进行遗传预测的可行性也已得到证实。虽然存在多种支持基于NGS的临床微生物学检测的生物信息学方法,但仍缺乏标准化、计算高效且可扩展的从样本到结果的工作流程,包括经过验证的质量控制参数。基于k-mer的生物信息学分析工作流程已被证明能够快速有效地分析从微生物测序应用中获得的大型基因组数据集。我们在此展示了基于k-mer的临床微生物学检测在全基因组测序(WGS)中的适用性,包括变异检测、分类鉴定、细菌分型以及AMR标记检测。湿实验室和干实验室工作流程是根据临床实验室改进法案(CLIA)关于实验室开发检测(LDT)的指南,针对多重耐药的ESKAPE病原体开发并验证的。通过日内和日间一式三份测量确定,所开发的基于k-mer的工作流程在变异检测和应用检测方面表现出≥99.39%的重复性、≥99.09%的再现性和≥99.76%的准确性。发现各检测方法的检测限在20×测序深度,AMR标记检测的检测限为15×。对基于k-mer的工作流程进行的全面基准测试表明,其分析性能标准与临床微生物学检测中基于比对的先进工作流程相当。两种方法对多位点序列分型(MLST)和系统发育分析的诊断敏感性和特异性均为100%。对于AMR标记检测,基于k-mer的工作流程的敏感性和特异性分别为95.29%和99.78%,而基于比对的方法的敏感性和特异性分别为95.17%和99.77%。综上所述,结果表明基于k-mer的分析工作流程能够实现广泛的临床微生物学检测,不仅可能用于基于WGS的分型和AMR基因检测,还能用于抗生素敏感性测试结果的遗传预测。