Transversal Activities in Applied Genomics, Sciensano, Brussels, Belgium.
Department of Plant Biotechnology and Bioinformatics, Ghent University, Ghent, Belgium.
J Clin Microbiol. 2021 May 19;59(6). doi: 10.1128/JCM.00202-21.
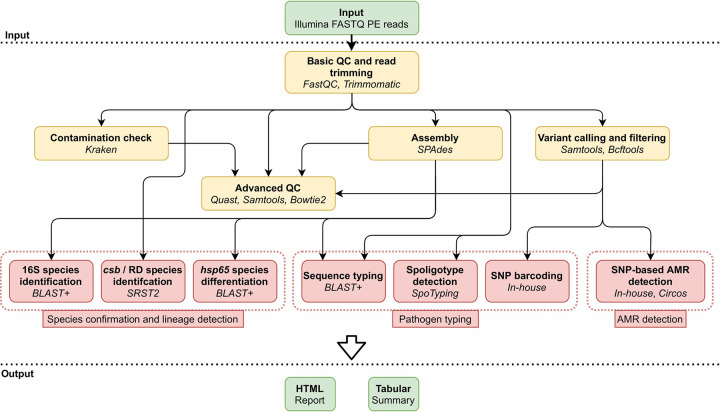
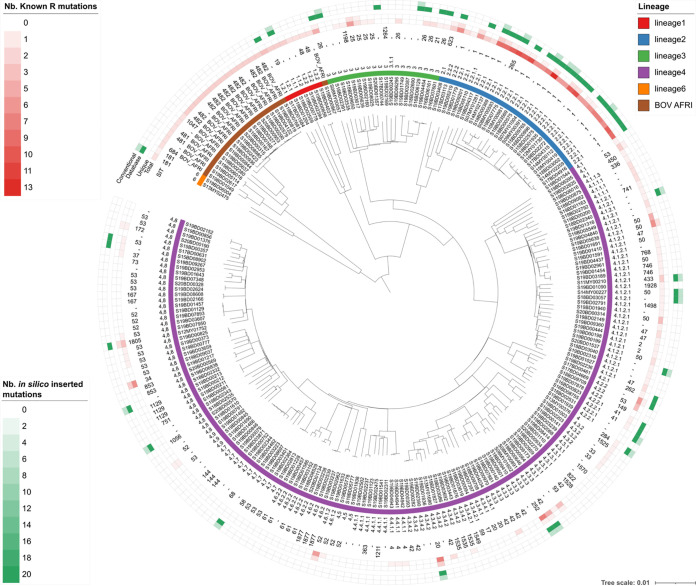
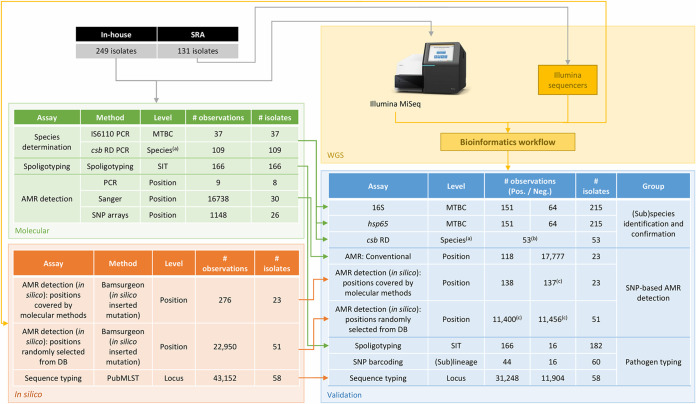
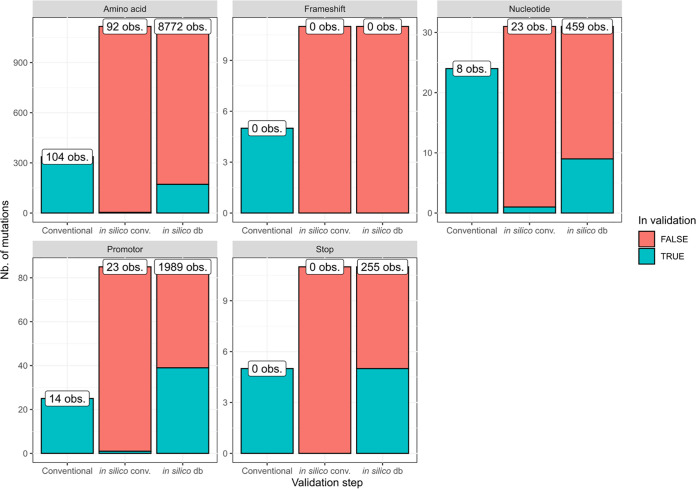
The use of whole-genome sequencing (WGS) for routine typing of bacterial isolates has increased substantially in recent years. For (MTB), in particular, WGS has the benefit of drastically reducing the time required to generate results compared to most conventional phenotypic methods. Consequently, a multitude of solutions for analyzing WGS MTB data have been developed, but their successful integration in clinical and national reference laboratories is hindered by the requirement for their validation, for which a consensus framework is still largely absent. We developed a bioinformatics workflow for (Illumina) WGS-based routine typing of MTB complex (MTBC) member isolates allowing complete characterization, including (sub)species confirmation and identification (16S, /RD, ), single nucleotide polymorphism (SNP)-based antimicrobial resistance (AMR) prediction, and pathogen typing (spoligotyping, SNP barcoding, and core genome multilocus sequence typing). Workflow performance was validated on a per-assay basis using a collection of 238 in-house-sequenced MTBC isolates, extensively characterized with conventional molecular biology-based approaches supplemented with public data. For SNP-based AMR prediction, results from molecular genotyping methods were supplemented with modified data sets, allowing us to greatly increase the set of evaluated mutations. The workflow demonstrated very high performance with performance metrics of >99% for all assays, except for spoligotyping, where sensitivity dropped to ∼90%. The validation framework for our WGS-based bioinformatics workflow can aid in the standardization of bioinformatics tools by the MTB community and other SNP-based applications regardless of the targeted pathogen(s). The bioinformatics workflow is available for academic and nonprofit use through the Galaxy instance of our institute at https://galaxy.sciensano.be.
近年来,全基因组测序(WGS)在细菌分离物的常规分型中的应用大大增加。对于结核分枝杆菌(MTB),特别是与大多数传统表型方法相比,WGS 大大缩短了生成结果所需的时间。因此,已经开发了许多用于分析 WGS MTB 数据的解决方案,但由于需要对其进行验证,因此其在临床和国家参考实验室中的成功整合受到阻碍,而对于验证,仍然缺乏共识框架。我们开发了一个基于 Illumina 的 WGS 用于结核分枝杆菌复合体(MTBC)成员分离物的常规分型的生物信息学工作流程,允许进行完整的特征描述,包括(亚种)确认和鉴定(16S,/ RD ,),基于单核苷酸多态性(SNP)的抗微生物药物耐药性(AMR)预测和病原体分型( spoligotyping , SNP 条形码和核心基因组多位点序列分型)。使用包含 238 个内部测序的 MTBC 分离物的集合,通过基于常规分子生物学方法的广泛特征描述,并辅以公共数据,在每个分析中验证了工作流程的性能。对于基于 SNP 的 AMR 预测,分子基因分型方法的结果补充了修改后的数据集,从而使我们能够大大增加评估的突变数量。该工作流程的性能指标非常高,所有分析的准确率均>99%,除 spoligotyping 外,灵敏度降至约 90%。我们的基于 WGS 的生物信息学工作流程的验证框架可以通过 MTB 社区和其他基于 SNP 的应用程序来帮助标准化生物信息学工具,而无需针对目标病原体(s)。该生物信息学工作流程可通过我们研究所的 Galaxy 实例在 https://galaxy.sciensano.be 上供学术和非营利组织使用。