Dipartimento Scienze Agrarie, Alimentari e Forestali, University of Palermo, 90128, Palermo, Italy.
Laboratoire des Productions Animales et Fourragères, Institut National de La Recherche Agronomique de Tunisie, Université de Carthage, 2049, Ariana, Tunisia.
Sci Rep. 2020 Sep 3;10(1):14522. doi: 10.1038/s41598-020-71375-2.
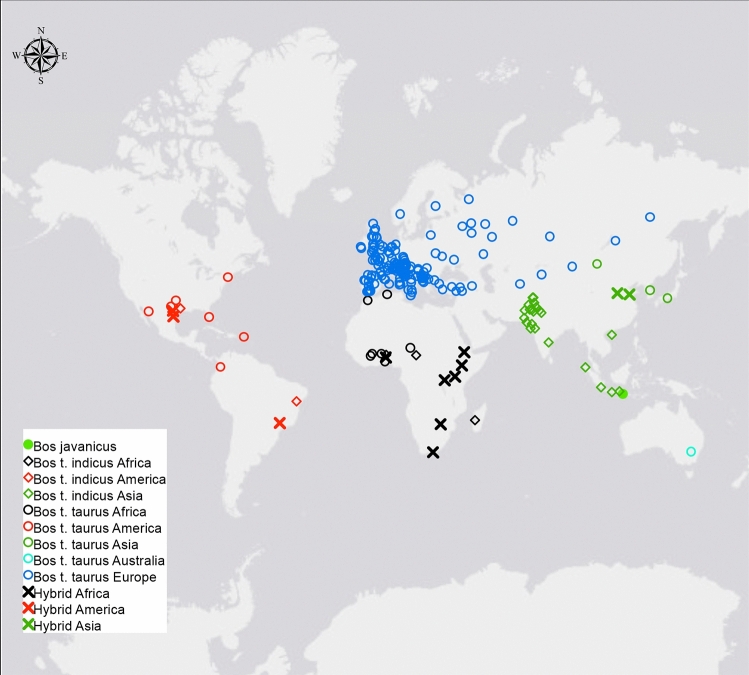
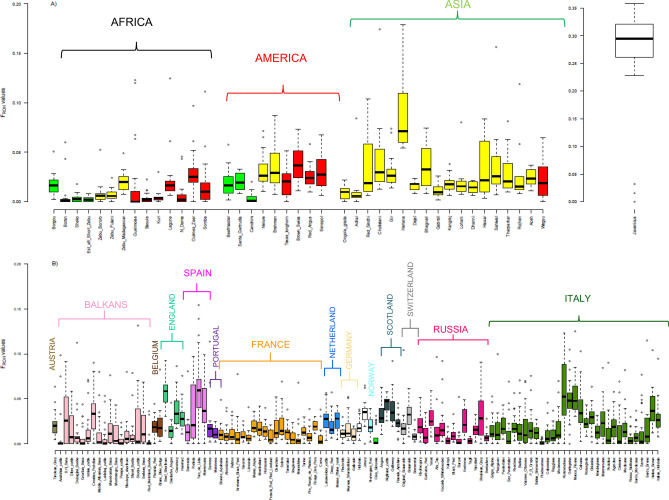
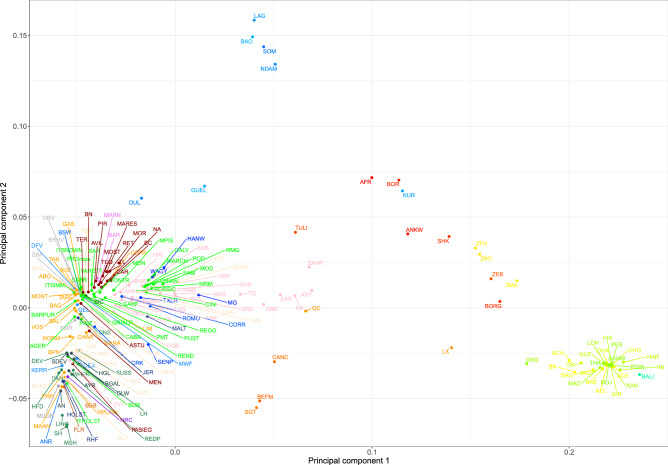
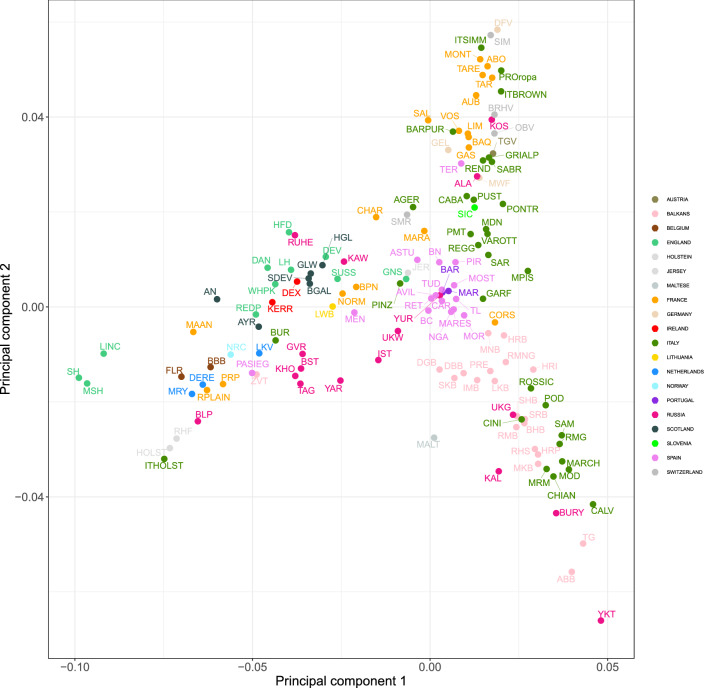
The availability of genotyping assays has allowed the detailed evaluation of cattle genetic diversity worldwide. However, these comprehensive studies did not include some local European populations, including autochthonous Italian cattle. In this study, we assembled a large-scale, genome-wide dataset of single nucleotide polymorphisms scored in 3,283 individuals from 205 cattle populations worldwide to assess genome-wide autozygosity and understand better the genetic relationships among these populations. We prioritized European cattle, with a special focus on Italian breeds. Moderate differences in estimates of molecular inbreeding calculated from runs of homozygosity (F) were observed among domesticated bovid populations from different geographic areas, except for Bali cattle. Our findings indicated that some Italian breeds show the highest estimates of levels of molecular inbreeding among the cattle populations assessed in this study. Patterns of genetic differentiation, shared ancestry, and phylogenetic analysis all suggested the occurrence of gene flow, particularly among populations originating from the same geographical area. For European cattle, we observed a distribution along three main directions, reflecting the known history and formation of the analyzed breeds. The Italian breeds are split into two main groups, based on their historical origin and degree of conservation of ancestral genomic components. The results pinpointed that also Sicilian breeds, much alike Podolian derived-breeds, in the past experienced a similar non-European influence, with African and indicine introgression.
基因分型检测的出现使得对全球范围内的牛种遗传多样性进行详细评估成为可能。然而,这些全面的研究并未包括一些欧洲当地群体,包括意大利本土牛种。在这项研究中,我们收集了来自全球 205 个牛种群的 3283 个个体的大规模全基因组单核苷酸多态性数据集,以评估全基因组近交程度,并更好地了解这些种群之间的遗传关系。我们特别关注欧洲牛种,其中重点是意大利品种。从同型交配系(F)计算的估计分子近交之间观察到适度的差异,这些估计是来自不同地理区域的驯化牛种群,巴厘牛除外。我们的研究结果表明,在这项研究评估的牛种群中,一些意大利品种显示出最高水平的分子近交程度。遗传分化、共同祖先和系统发育分析的模式都表明存在基因流动,特别是在来自同一地理区域的种群之间。对于欧洲牛种,我们观察到了沿着三个主要方向的分布,这反映了分析品种的已知历史和形成。意大利品种根据其历史起源和祖先基因组成分的保护程度分为两个主要群体。研究结果指出,西西里品种也与源自波多黎各的品种类似,过去经历了类似的非欧洲影响,存在非洲和印度血统的渗入。