Department of Computer Science, Johns Hopkins University, Baltimore, Maryland 21211, USA.
Cold Spring Harbor Laboratory, Cold Spring Harbor, New York 11724, USA.
Genome Res. 2020 Sep;30(9):1258-1273. doi: 10.1101/gr.260497.119. Epub 2020 Sep 4.
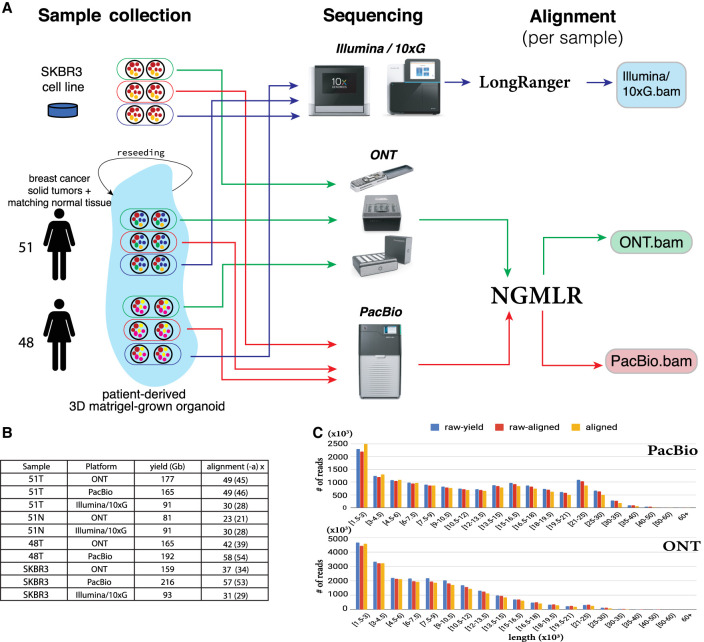
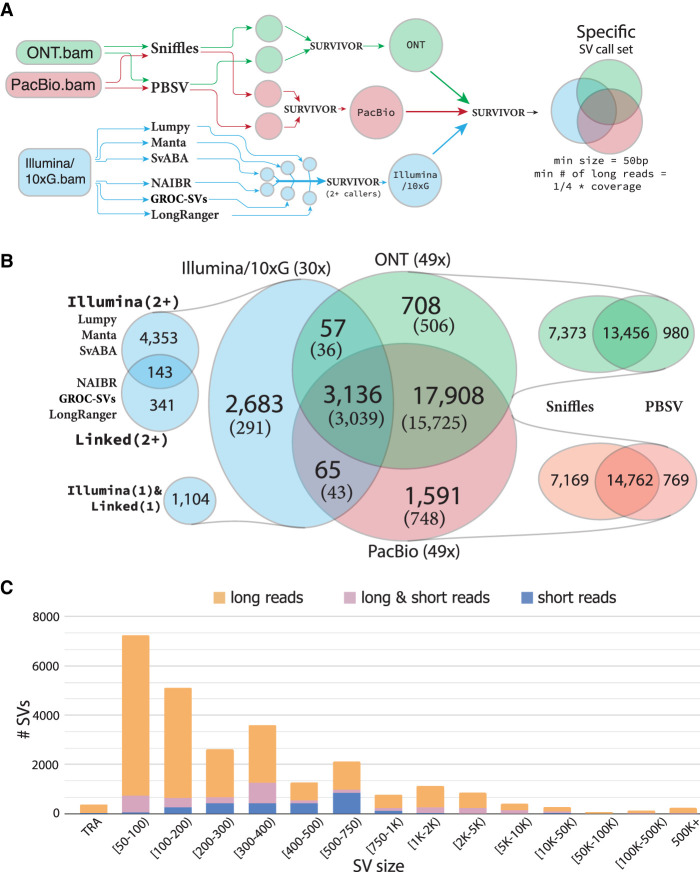
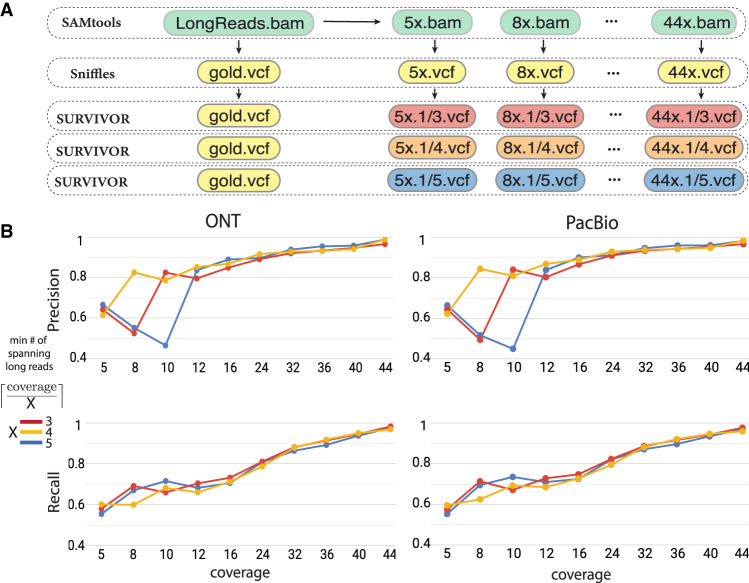
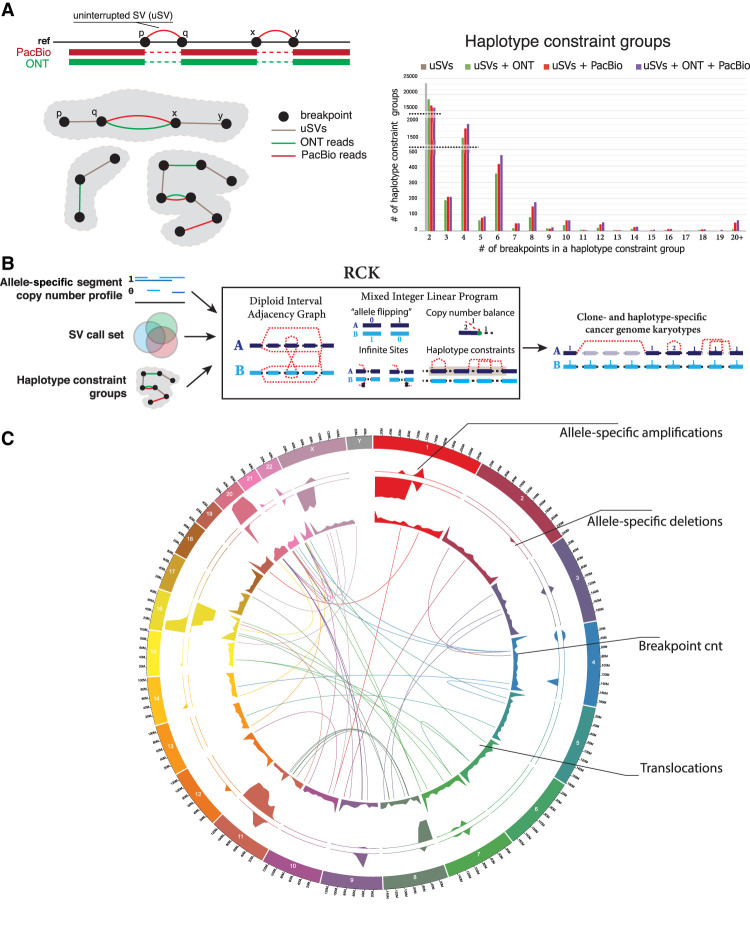
Improved identification of structural variants (SVs) in cancer can lead to more targeted and effective treatment options as well as advance our basic understanding of the disease and its progression. We performed whole-genome sequencing of the SKBR3 breast cancer cell line and patient-derived tumor and normal organoids from two breast cancer patients using Illumina/10x Genomics, Pacific Biosciences (PacBio), and Oxford Nanopore Technologies (ONT) sequencing. We then inferred SVs and large-scale allele-specific copy number variants (CNVs) using an ensemble of methods. Our findings show that long-read sequencing allows for substantially more accurate and sensitive SV detection, with between 90% and 95% of variants supported by each long-read technology also supported by the other. We also report high accuracy for long reads even at relatively low coverage (25×-30×). Furthermore, we integrated SV and CNV data into a unifying karyotype-graph structure to present a more accurate representation of the mutated cancer genomes. We find hundreds of variants within known cancer-related genes detectable only through long-read sequencing. These findings highlight the need for long-read sequencing of cancer genomes for the precise analysis of their genetic instability.
提高癌症结构变异(SVs)的识别能力可以为更有针对性和更有效的治疗方案提供依据,也有助于推进我们对疾病及其进展的基本认识。我们使用 Illumina/10x Genomics、Pacific Biosciences(PacBio)和 Oxford Nanopore Technologies(ONT)测序技术,对 SKBR3 乳腺癌细胞系和来自两名乳腺癌患者的肿瘤和正常类器官进行了全基因组测序。然后,我们使用一组方法推断 SV 和大规模等位基因特异性拷贝数变异(CNVs)。我们的研究结果表明,长读长测序可以更准确、更灵敏地检测 SV,每种长读长技术支持的变异中有 90%至 95%也得到了其他技术的支持。即使在相对较低的覆盖度(25×-30×)下,长读长的准确性也很高。此外,我们将 SV 和 CNV 数据整合到一个统一的核型图结构中,以更准确地呈现突变癌症基因组。我们发现,只有通过长读长测序才能检测到数百个已知与癌症相关的基因中的变异。这些发现强调了需要对癌症基因组进行长读长测序,以精确分析其遗传不稳定性。