Department of Chemistry, University of Jordan, Amman, 11942, Jordan.
Department of Chemistry, Memorial University, St. John's, NL, A1B 3X7, Canada.
Sci Rep. 2020 Sep 14;10(1):15025. doi: 10.1038/s41598-020-71881-3.
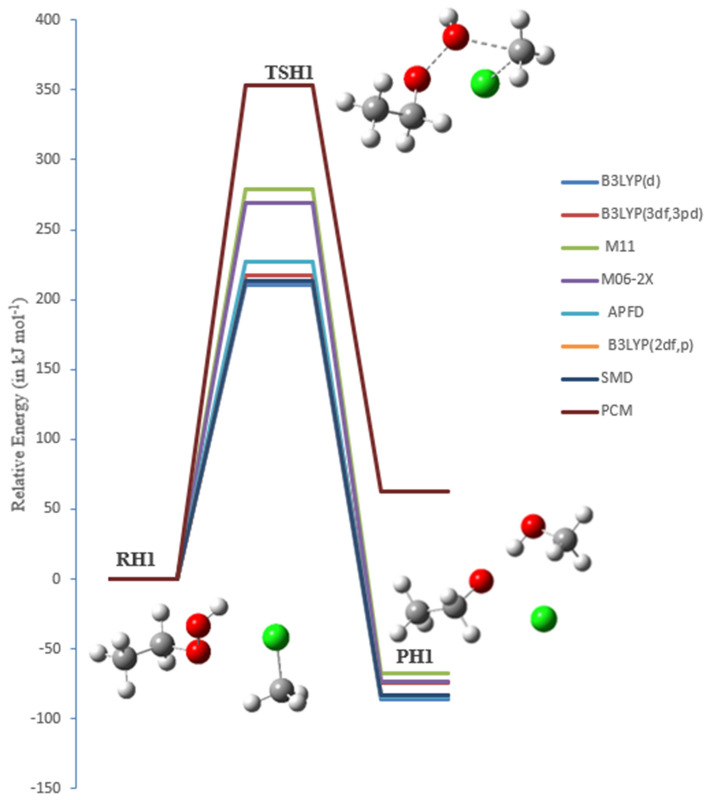
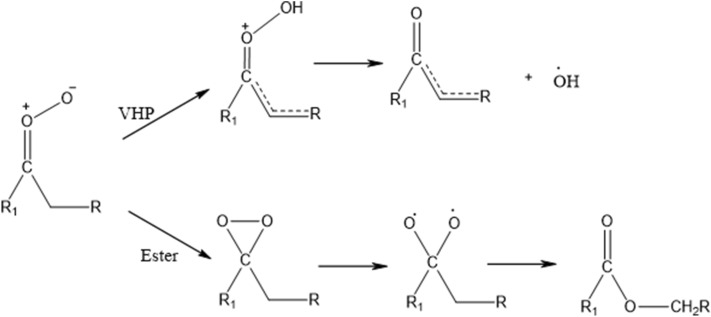
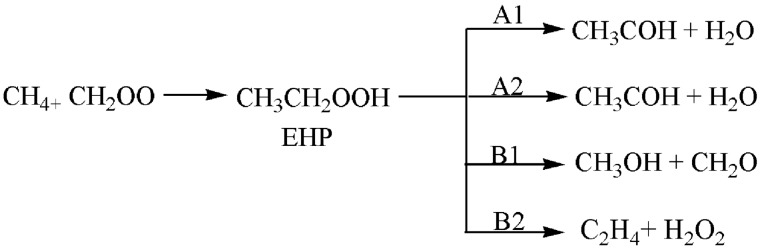
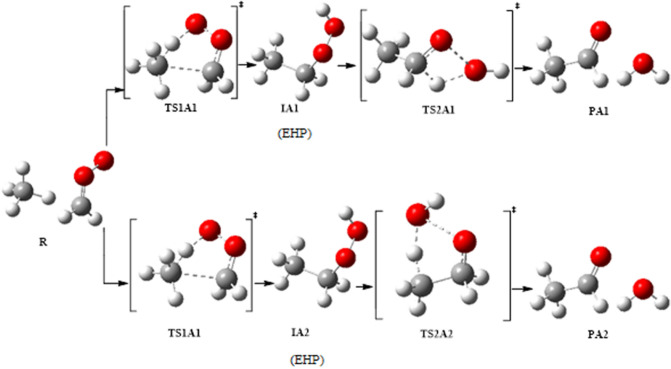
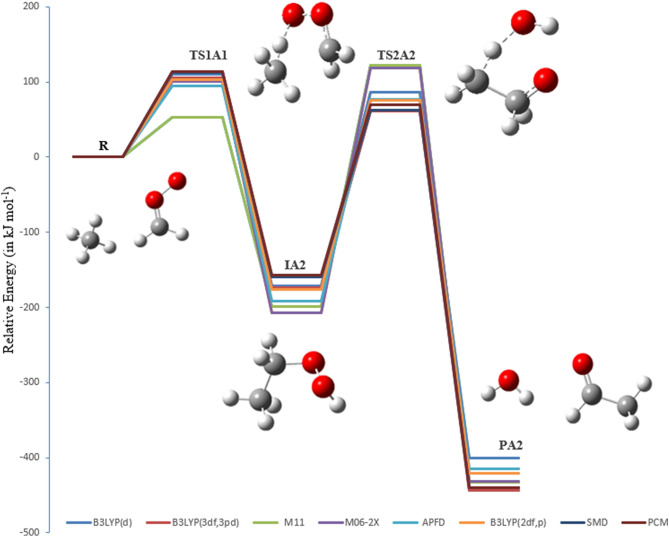
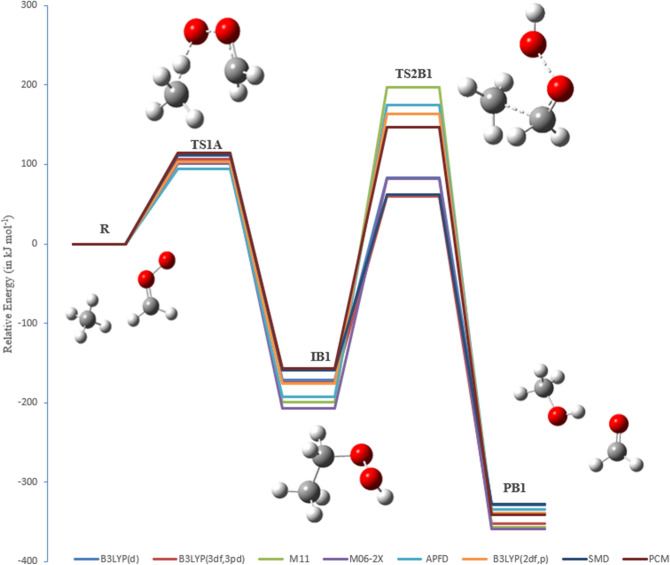
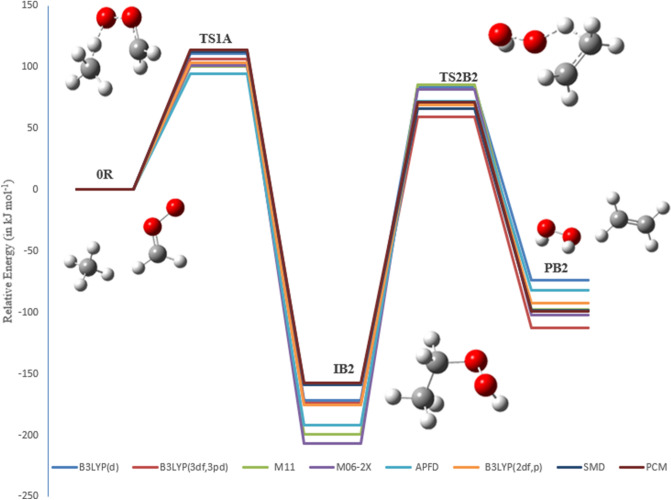
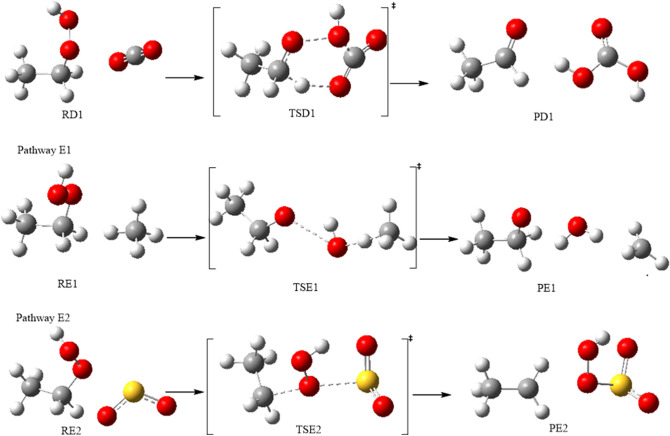
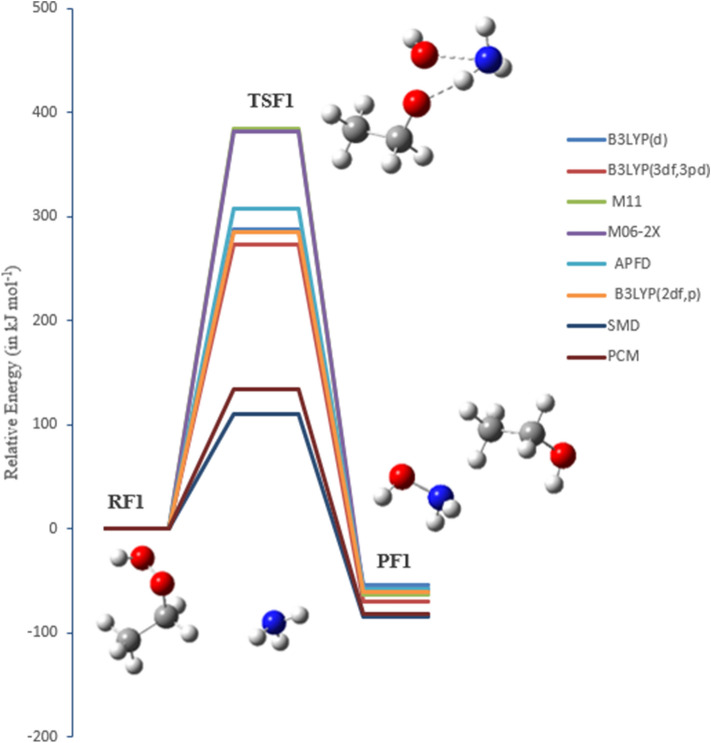
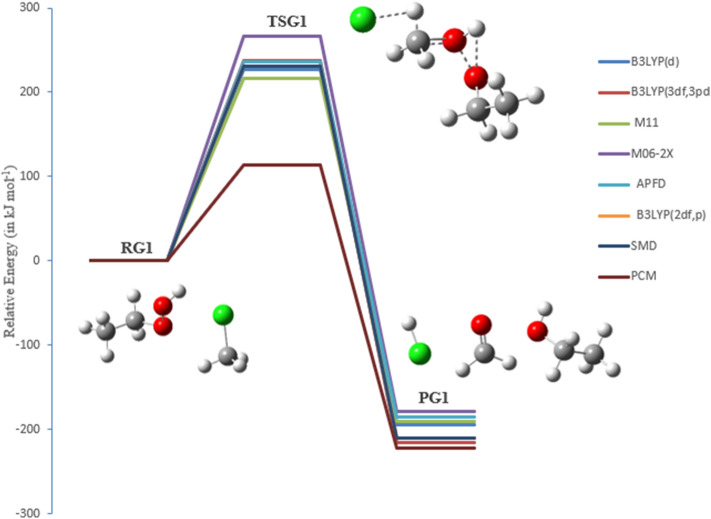
A detailed computational study of the atmospheric reaction of the simplest Criegee intermediate CHOO with methane has been performed using the density functional theory (DFT) method and high-level calculations. Solvation models were utilized to address the effect of water molecules on prominent reaction steps and their associated energies. The structures of all proposed mechanisms were optimized using B3LYP functional with several basis sets: 6-31G(d), 6-31G (2df,p), 6-311++G(3df,3pd) and at M06-2X/6-31G(d) and APFD/6-31G(d) levels of theory. Furthermore, all structures were optimized at the B3LYP/6-311++G(3df,3pd) level of theory. The intrinsic reaction coordinate (IRC) analysis was performed for characterizing the transition states on the potential energy surfaces. Fifteen different mechanistic pathways were studied for the reaction of Criegee intermediate with methane. Both thermodynamic functions (ΔH and ΔG), and activation parameters (activation energies E, enthalpies of activation ΔH, and Gibbs energies of activation ΔG) were calculated for all pathways investigated. The individual mechanisms for pathways A1, A2, B1, and B2, comprise two key steps: (i) the formation of ethyl hydroperoxide (EHP) accompanying with the hydrogen transfer from the alkanes to the terminal oxygen atom of CIs, and (ii) a following unimolecular dissociation of EHP. Pathways from C1 → H1 involve the bimolecular reaction of EHP with different atmospheric species. The photochemical reaction of methane with EHP (pathway E1) was found to be the most plausible reaction mechanism, exhibiting an overall activation energy of 7 kJ mol, which was estimated in vacuum at the B3LYP/6-311++G(3df,3pd) level of theory. All of the reactions were found to be strongly exothermic, expect the case of the sulfur dioxide-involved pathway that is predicted to be endothermic. The solvent effect plays an important role in the reaction of EHP with ammonia (pathway F1). Compared with the gas phase reaction, the overall activation energy for the solution phase reaction is decreased by 162 and 140 kJ mol according to calculations done with the SMD and PCM solvation models, respectively.
采用密度泛函理论(DFT)方法和高精度计算对最简单的 Criegee 中间体 CHOO 与甲烷的大气反应进行了详细的计算研究。利用溶剂化模型来解决水分子对显著反应步骤及其相关能量的影响。使用 B3LYP 函数和几种基组(6-31G(d)、6-31G(2df,p)、6-311++G(3df,3pd)和 M06-2X/6-31G(d)和 APFD/6-31G(d))对所有建议的机制的结构进行了优化。此外,所有结构均在 B3LYP/6-311++G(3df,3pd)理论水平下进行优化。通过内禀反应坐标(IRC)分析对势能面上的过渡态进行了特征化。研究了 Criegee 中间体与甲烷反应的 15 种不同的反应途径。所有研究途径的热力学函数(ΔH 和 ΔG)和活化参数(活化能 E、活化焓ΔH 和活化吉布斯能ΔG)均进行了计算。途径 A1、A2、B1 和 B2 的各个机制包括两个关键步骤:(i)乙基过氧化物(EHP)的形成,同时伴随着烷烃向 CIs 末端氧原子的氢转移,以及(ii)EHP 的后续单分子分解。途径 C1→H1 涉及 EHP 与不同大气物质的双分子反应。在真空条件下,通过 B3LYP/6-311++G(3df,3pd)理论水平估算,甲烷与 EHP 的光化学反应(途径 E1)被认为是最合理的反应机制,其总活化能为 7 kJ mol。所有反应均被发现为强放热反应,而涉及二氧化硫的反应情况则预计为吸热反应。溶剂效应在 EHP 与氨的反应(途径 F1)中起着重要作用。与气相反应相比,根据 SMD 和 PCM 溶剂化模型的计算,溶液相反应的总活化能分别降低了 162 和 140 kJ mol。