Department of Medicine I, University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
Leibniz Research Centre for Working Environment and Human Factors, Technical University Dortmund, Dortmund, Germany; Department of Forensic Medicine and Toxicology, Faculty of Veterinary Medicine, South Valley University, Qena, Egypt.
Cell Mol Gastroenterol Hepatol. 2021;11(2):371-388. doi: 10.1016/j.jcmgh.2020.09.002. Epub 2020 Sep 12.
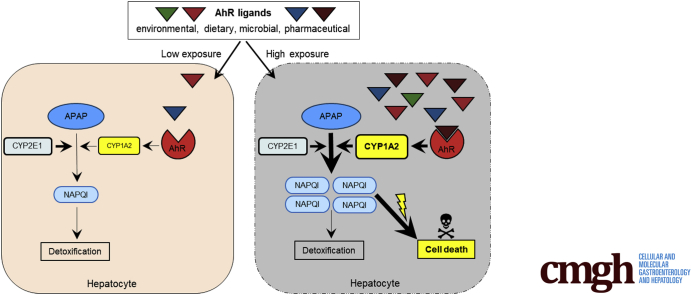
BACKGROUND & AIMS: Acetaminophen (APAP)-induced liver injury is one of the most common causes of acute liver failure, however, a clear definition of sensitizing risk factors is lacking. Here, we investigated the role of the ligand-activated transcription factor aryl hydrocarbon receptor (Ahr) in APAP-induced liver injury. We hypothesized that Ahr, which integrates environmental, dietary, microbial and metabolic signals into complex cellular transcriptional programs, might act as a rheostat for APAP-toxicity.
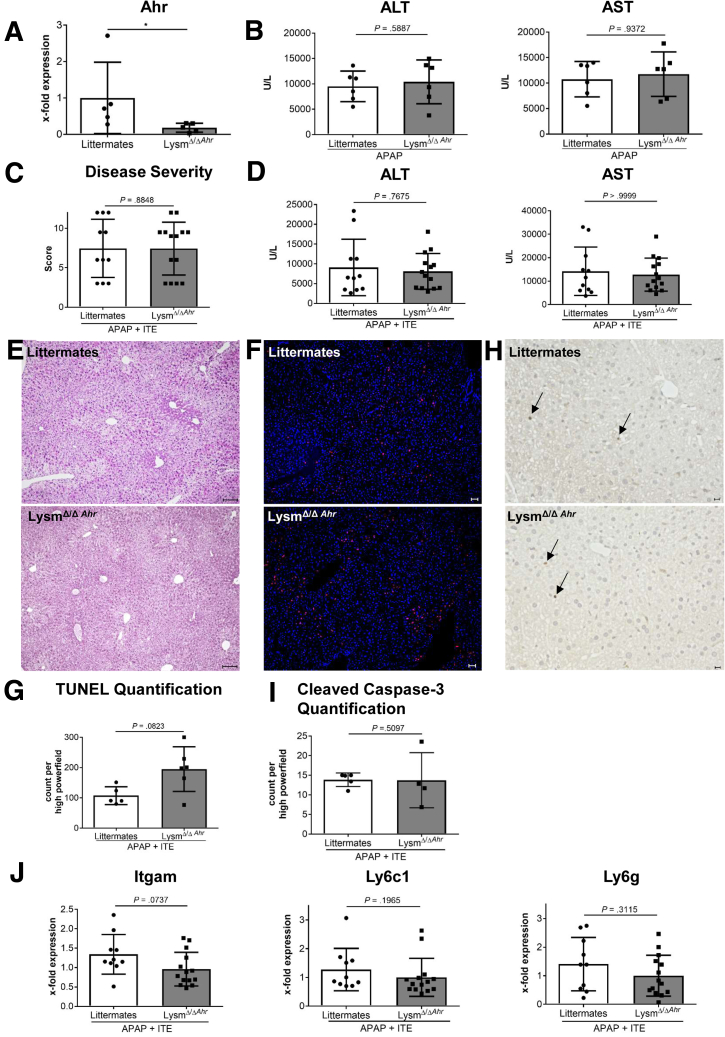
Wildtype or conditional Ahr knockout mice lacking Ahr in hepatocytes (Alb) or myeloid cells (LysM) were treated with the specific Ahr ligand 2-(1'H-indole-3'-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) together with APAP.
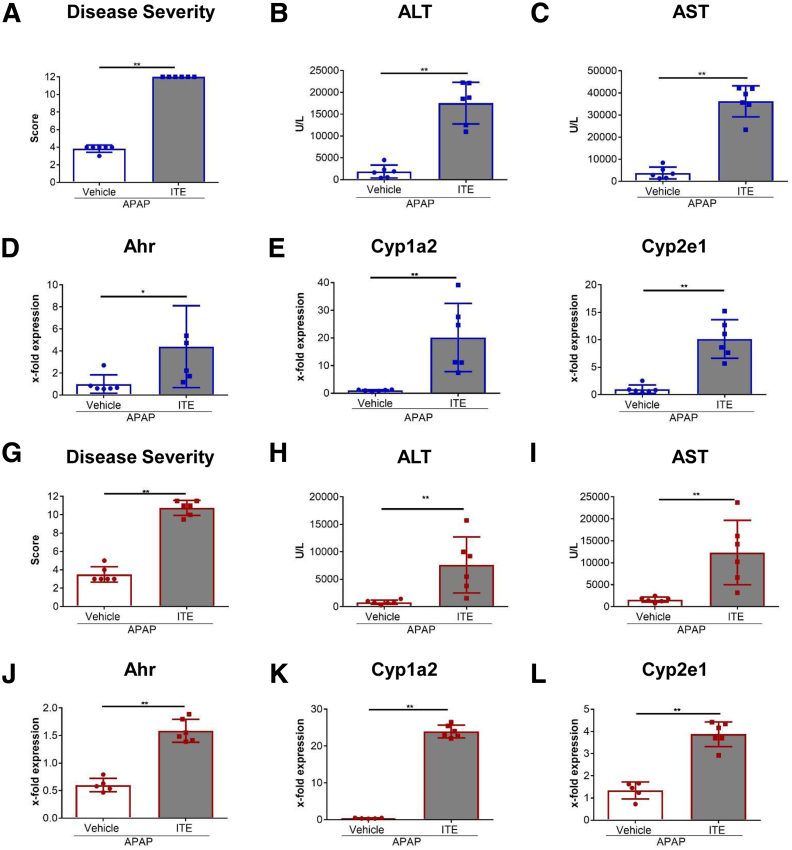
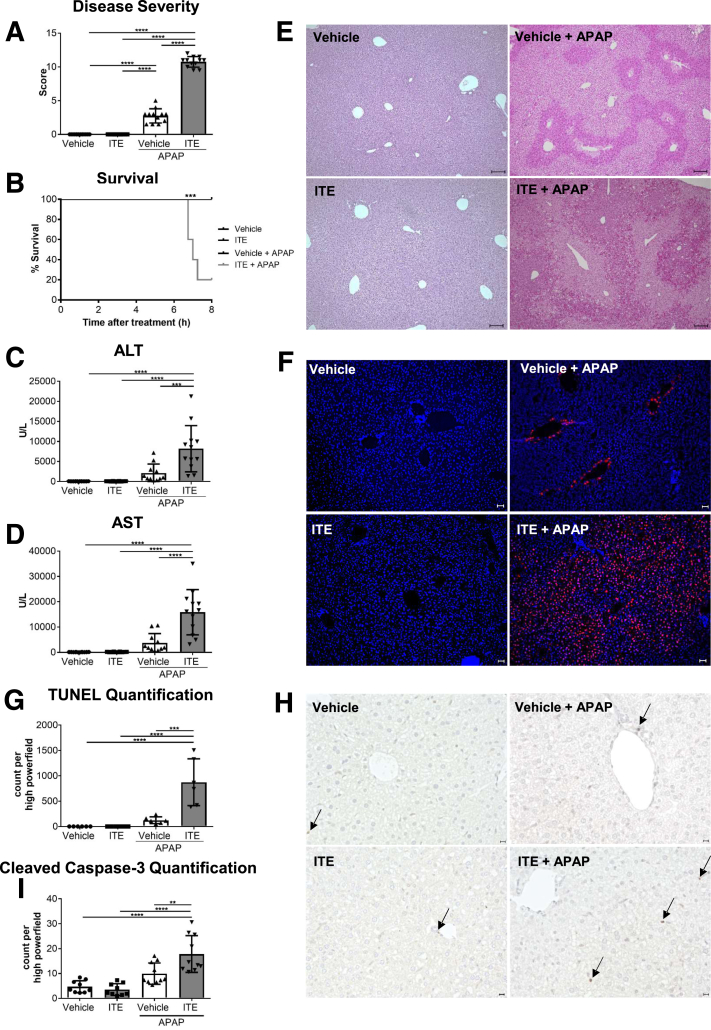
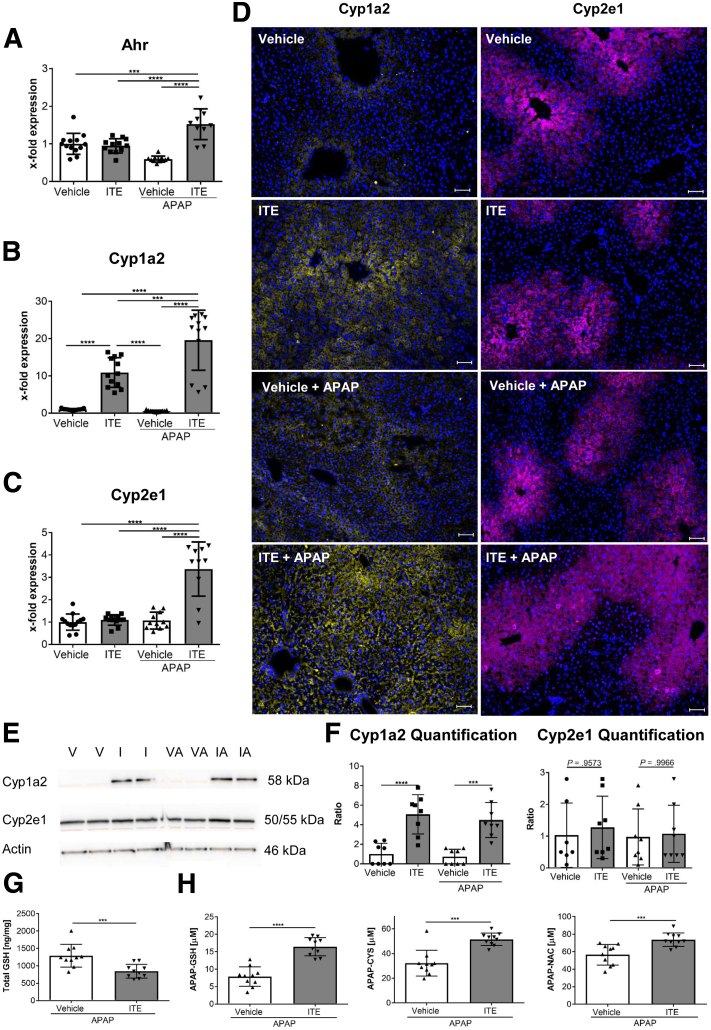
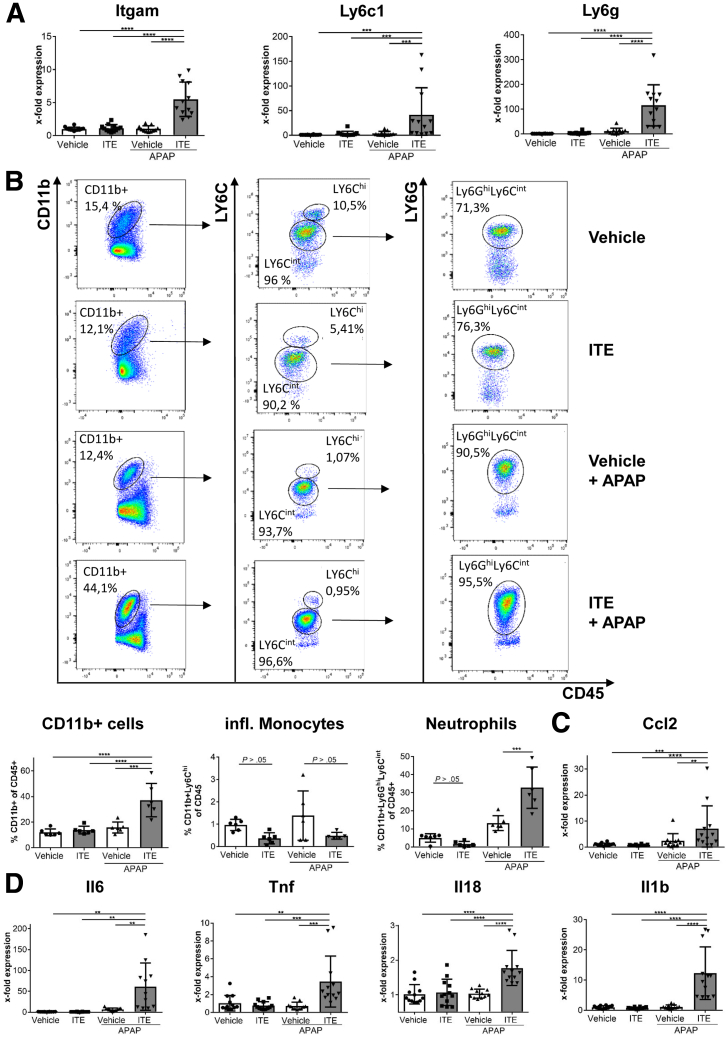
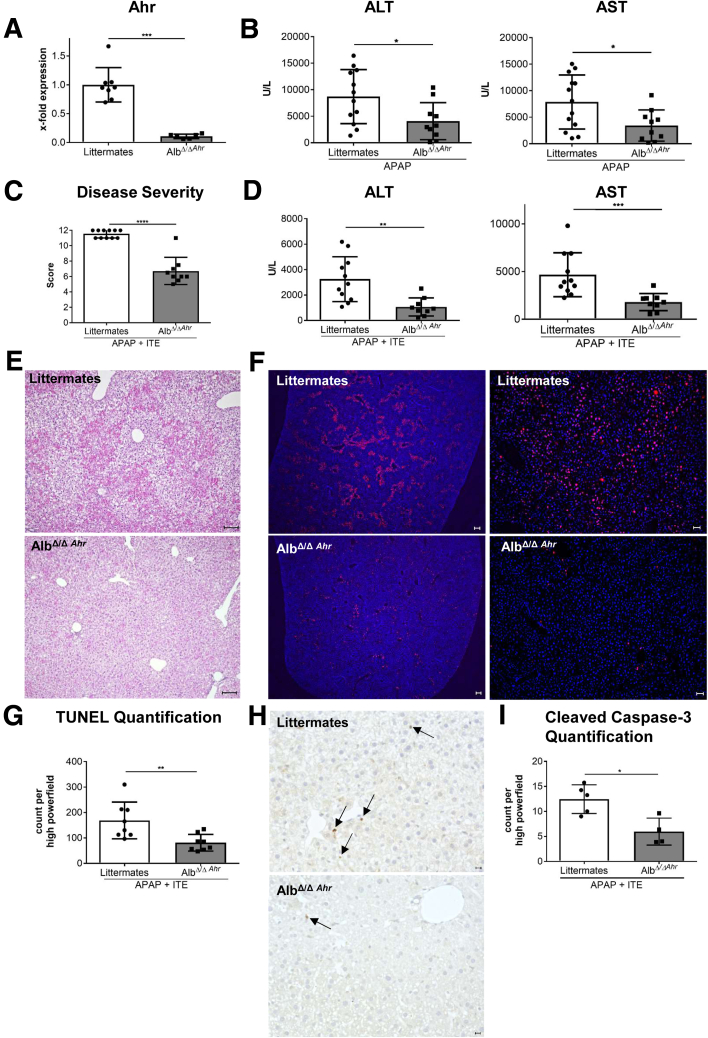
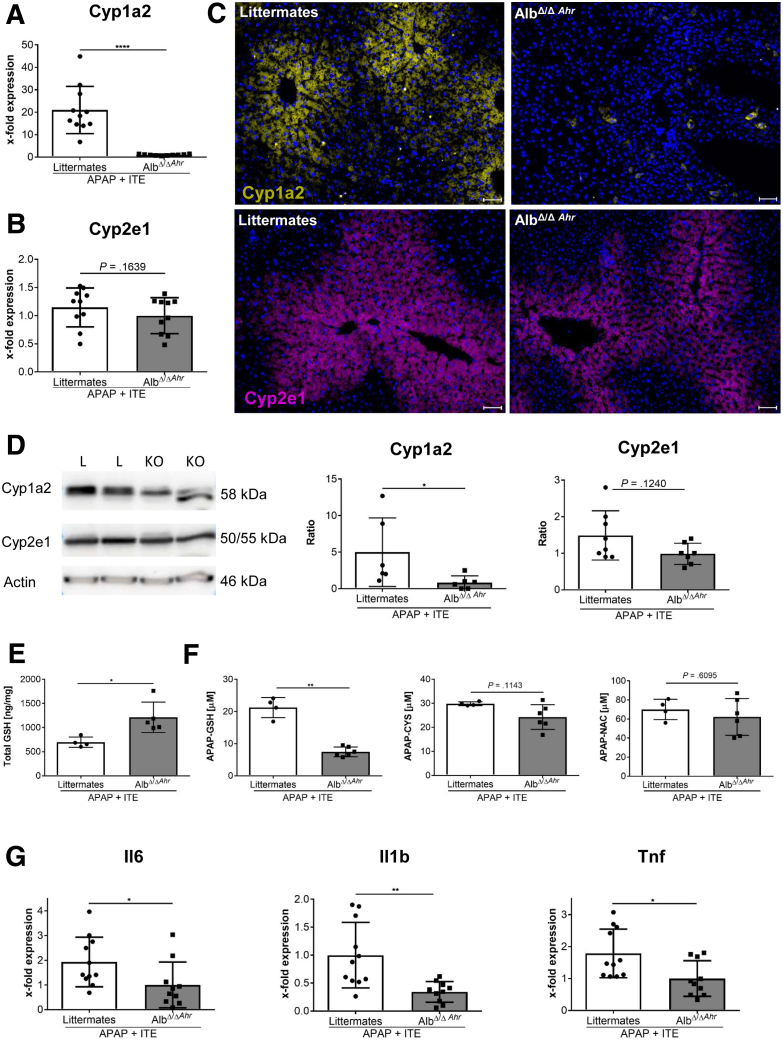
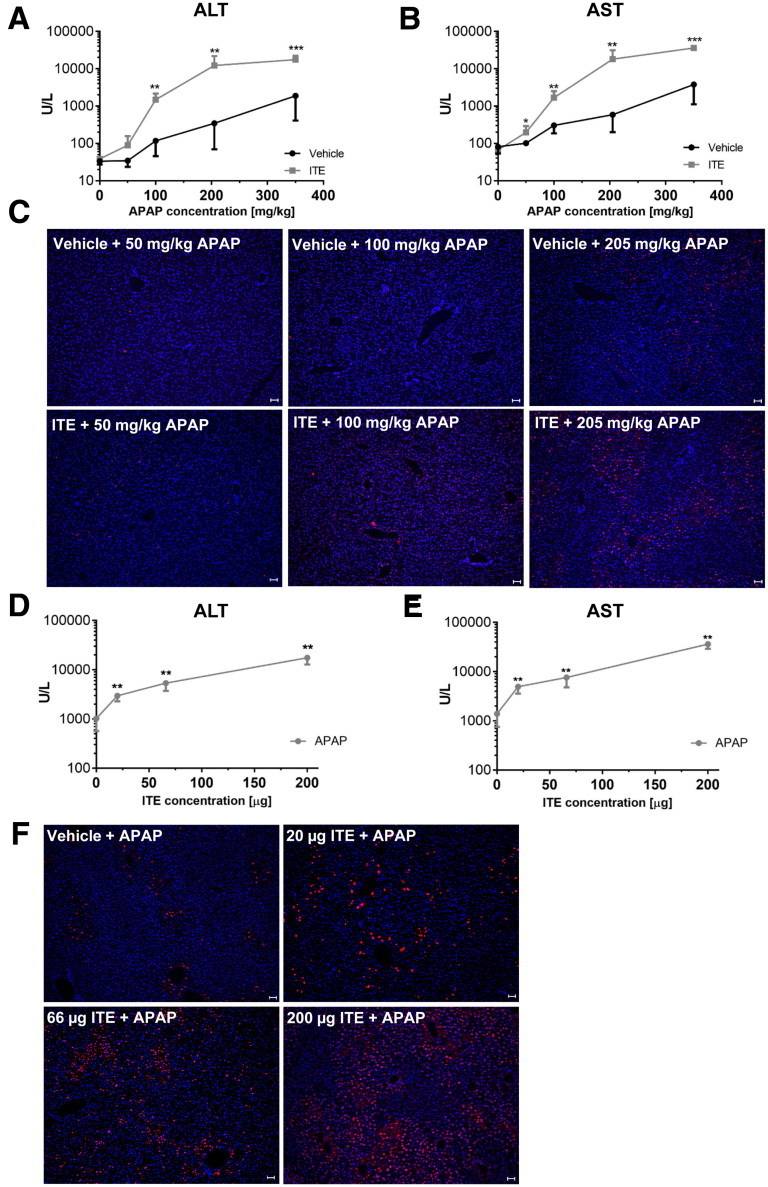
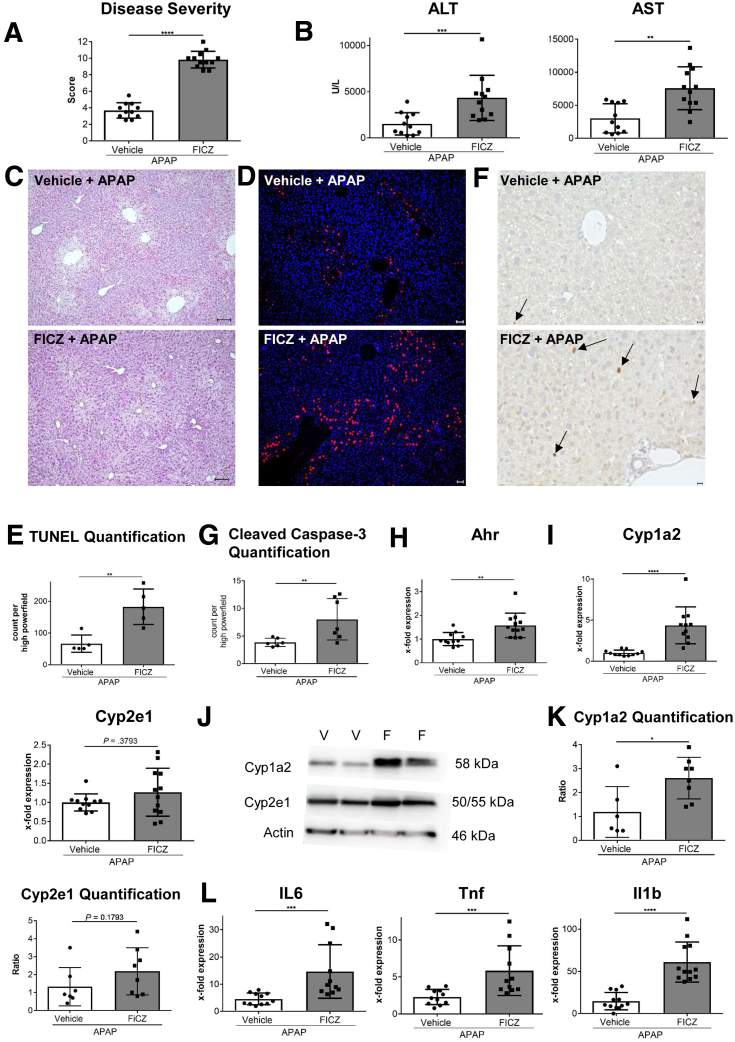
Ahr activation by ITE, which by itself was non-toxic, exacerbated APAP-induced hepatotoxicity compared to vehicle-treated controls, causing 80% vs. 0% mortality after administration of a normally sublethal APAP overdose. Of note, Ahr activation induced hepatocyte death even at APAP doses within the therapeutic range. Aggravated liver injury was associated with significant neutrophil infiltration; however, lack of Ahr in myeloid cells did not protect LysM mice from exacerbated APAP hepatotoxicity. In contrast, Alb mice were largely protected from ITE-induced aggravated liver damage, indicating that Ahr activation in hepatocytes, but not in myeloid cells, was instrumental for disease exacerbation. Mechanistically, Ahr activation fueled hepatic accumulation of toxic APAP metabolites by up-regulating expression of the APAP-metabolizing enzyme Cyp1a2, a direct Ahr downstream target.
Ahr activation in hepatocytes potentiates APAP-induced hepatotoxicity. Thus, individual exposition to environmental Ahr ligands might explain individual sensitivity to hyperacute liver failure.
对乙酰氨基酚(APAP)诱导的肝损伤是急性肝衰竭最常见的原因之一,但缺乏明确的致敏危险因素定义。在这里,我们研究了配体激活转录因子芳香烃受体(Ahr)在 APAP 诱导的肝损伤中的作用。我们假设,Ahr 可以将环境、饮食、微生物和代谢信号整合到复杂的细胞转录程序中,可能作为 APAP 毒性的变阻器。
用特异性 Ahr 配体 2-(1'H-吲哚-3'-羰基)-噻唑-4-羧酸甲酯(ITE)联合 APAP 处理野生型或条件性 Ahr 敲除小鼠(肝细胞缺乏 Ahr [Alb]或髓系细胞缺乏 Ahr [LysM])。
ITE 激活 Ahr(本身无毒性)可加剧 APAP 诱导的肝毒性,与用载体处理的对照组相比,导致给予正常亚致死剂量 APAP 过量后 80% vs. 0%的死亡率。值得注意的是,即使在 APAP 剂量处于治疗范围内,Ahr 激活也会诱导肝细胞死亡。加重的肝损伤与显著的中性粒细胞浸润有关;然而,髓系细胞中缺乏 Ahr 并不能保护 LysM 小鼠免受加重的 APAP 肝毒性。相反,Alb 小鼠在很大程度上免受 ITE 诱导的加重的肝损伤的保护,表明 Ahr 在肝细胞中的激活,而不是在髓系细胞中的激活,是疾病加重的关键。从机制上讲,Ahr 激活通过上调 APAP 代谢酶 Cyp1a2 的表达来促进肝内有毒的 APAP 代谢物的积累,Cyp1a2 是 Ahr 的直接下游靶标。
肝细胞中 Ahr 的激活增强了 APAP 诱导的肝毒性。因此,个体暴露于环境 Ahr 配体可能解释了个体对急性肝衰竭的敏感性。