Translational Neurodegeneration Section "Albrecht-Kossel", Department of Neurology, University Medical Center Rostock, University of Rostock, 18147 Rostock, Germany.
Center for Regenerative Therapies Dresden (CRTD), Technische Universität Dresden, Dresden, Germany.
Int J Mol Sci. 2020 Sep 21;21(18):6938. doi: 10.3390/ijms21186938.
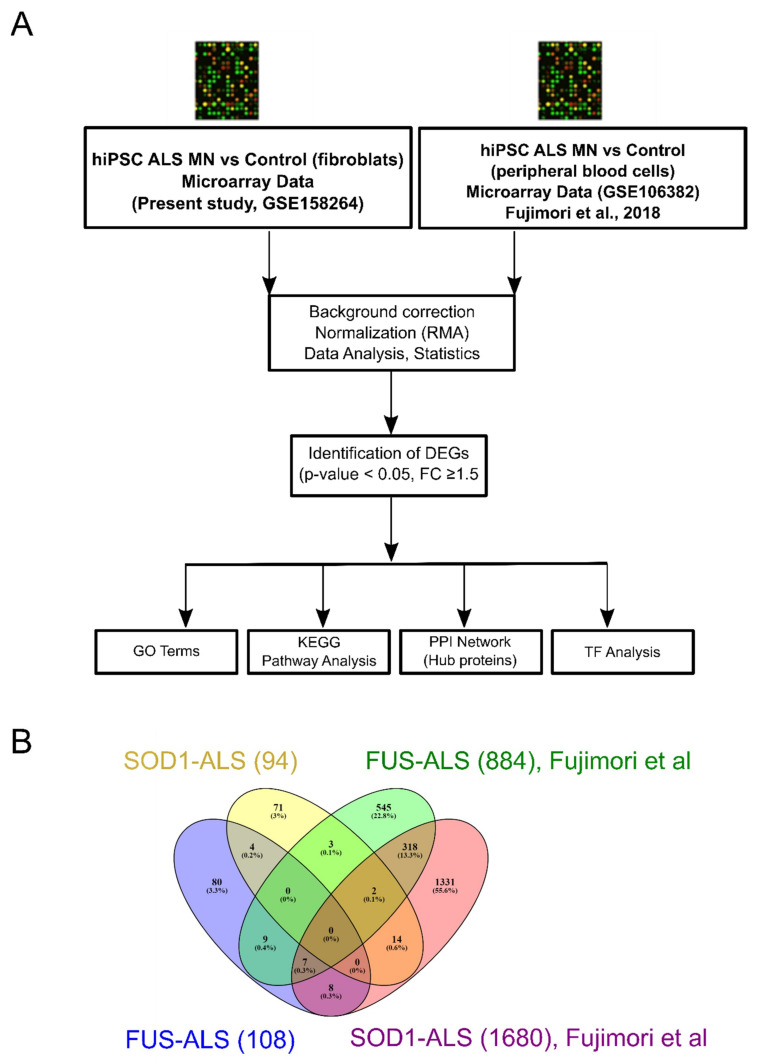
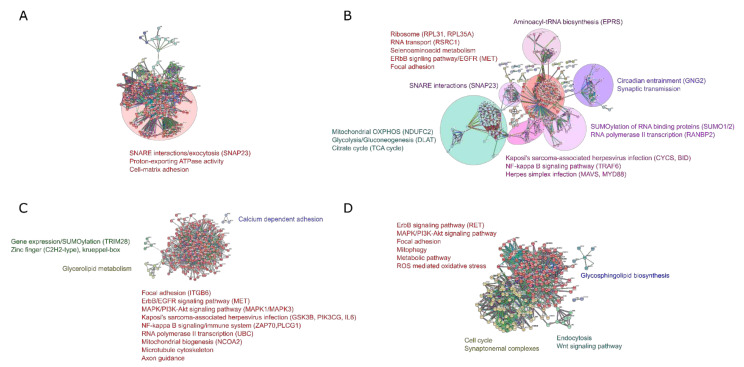
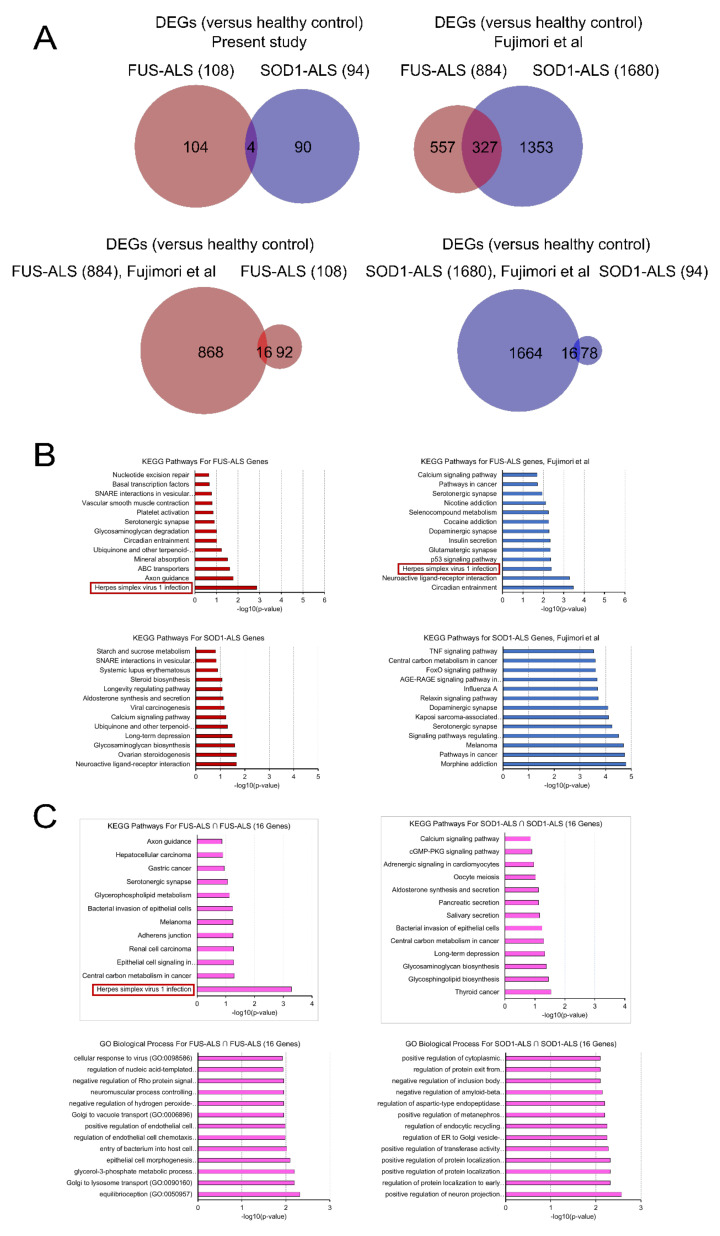
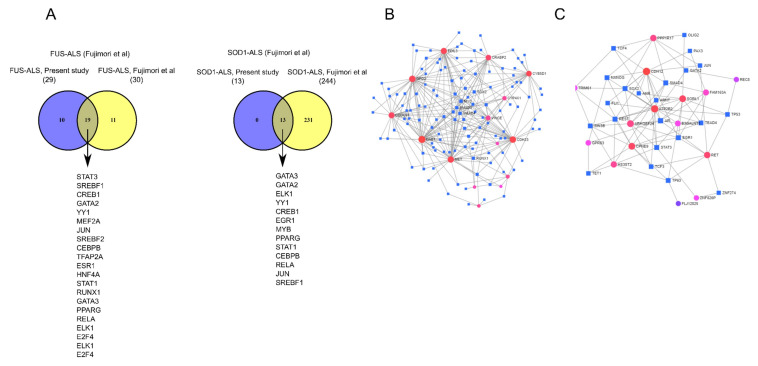
Amyotropic lateral sclerosis (ALS) is a lethally progressive and irreversible neurodegenerative disease marked by apparent death of motor neurons present in the spinal cord, brain stem and motor cortex. While more and more gene mutants being established for genetic ALS, the vast majority suffer from sporadic ALS (>90%). It has been challenging, thus, to model sporadic ALS which is one reason why the underlying pathophysiology remains elusive and has stalled the development of therapeutic strategies of this progressive motor neuron disease. To further unravel these pathological signaling pathways, human induced pluripotent stem cell (hiPSCs)-derived motor neurons (MNs) from FUS- and SOD1 ALS patients and healthy controls were systematically compared to independent published datasets. Here through this study we created a gene profile of ALS by analyzing the DEGs, the Kyoto encyclopedia of Genes and Genomes (KEGG) pathways, the interactome and the transcription factor profiles (TF) that would identify altered molecular/functional signatures and their interactions at both transcriptional (mRNAs) and translational levels (hub proteins and TFs). Our findings suggest that FUS and SOD1 may develop from dysregulation in several unique pathways and herpes simplex virus (HSV) infection was among the topmost predominant cellular pathways connected to FUS and not to SOD1. In contrast, SOD1 is mainly characterized by alterations in the metabolic pathways and alterations in the neuroactive-ligand-receptor interactions. This suggests that different genetic ALS forms are singular diseases rather than part of a common spectrum. This is important for patient stratification clearly pointing towards the need for individualized medicine approaches in ALS.
肌萎缩侧索硬化症(ALS)是一种致命的进行性和不可逆转的神经退行性疾病,其特征是脊髓、脑干和运动皮层中的运动神经元明显死亡。虽然越来越多的基因突变为遗传性 ALS 所确立,但绝大多数患有散发性 ALS(>90%)。因此,模拟散发性 ALS 一直具有挑战性,这也是导致其潜在病理生理学仍难以捉摸并阻碍了这种进行性运动神经元疾病治疗策略发展的原因之一。为了进一步揭示这些病理信号通路,我们系统地比较了来自 FUS 和 SOD1 ALS 患者和健康对照的人类诱导多能干细胞(hiPSC)衍生的运动神经元(MN)与独立发表的数据集。通过这项研究,我们通过分析差异表达基因(DEGs)、京都基因与基因组百科全书(KEGG)通路、相互作用组和转录因子谱(TF),创建了 ALS 的基因谱,这些分析可以确定在转录水平(mRNA)和翻译水平(枢纽蛋白和 TF)上改变的分子/功能特征及其相互作用。我们的研究结果表明,FUS 和 SOD1 可能是由于几个独特通路的失调而发展而来,单纯疱疹病毒(HSV)感染是与 FUS 而非 SOD1 相关的最主要的细胞通路之一。相比之下,SOD1 主要表现为代谢通路的改变和神经活性配体-受体相互作用的改变。这表明不同的遗传性 ALS 形式是独立的疾病,而不是共同谱的一部分。这对于患者分层非常重要,明确指出需要在 ALS 中采用个体化医学方法。