Department of Biotechnology and Genetic Engineering, Faculty of Life Science, Mawlana Bhashani Science and Technology University, Tangail-1902, Bangladesh; Aristopharma Limited, Bangladesh.
Department of Biotechnology and Genetic Engineering, Faculty of Life Science, Mawlana Bhashani Science and Technology University, Tangail-1902, Bangladesh; Department of Medical Biotechnology, Bangladesh University of Health Sciences, Dhaka, Bangladesh.
Int J Antimicrob Agents. 2020 Dec;56(6):106177. doi: 10.1016/j.ijantimicag.2020.106177. Epub 2020 Sep 25.
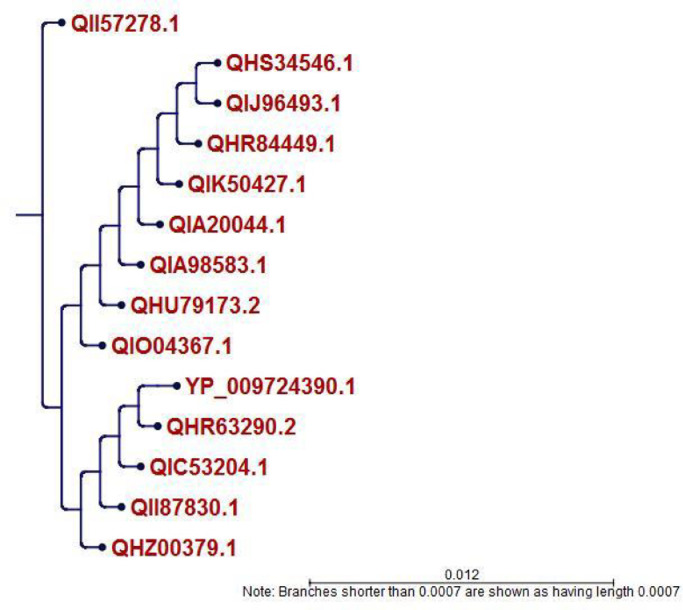
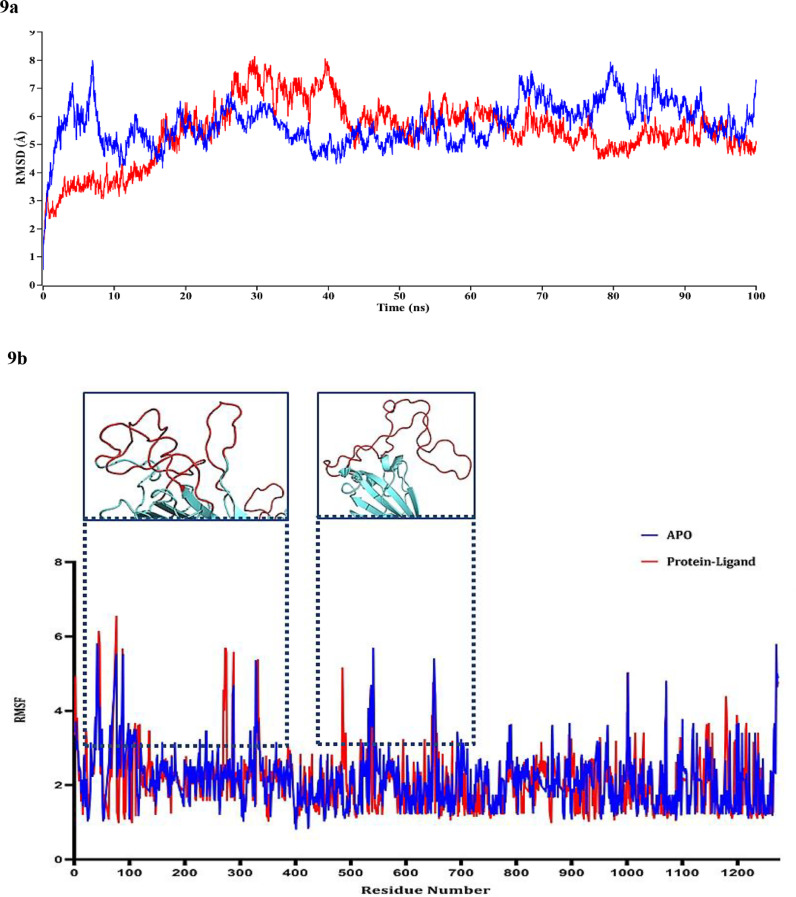
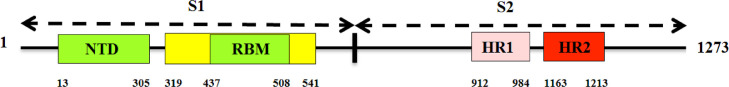
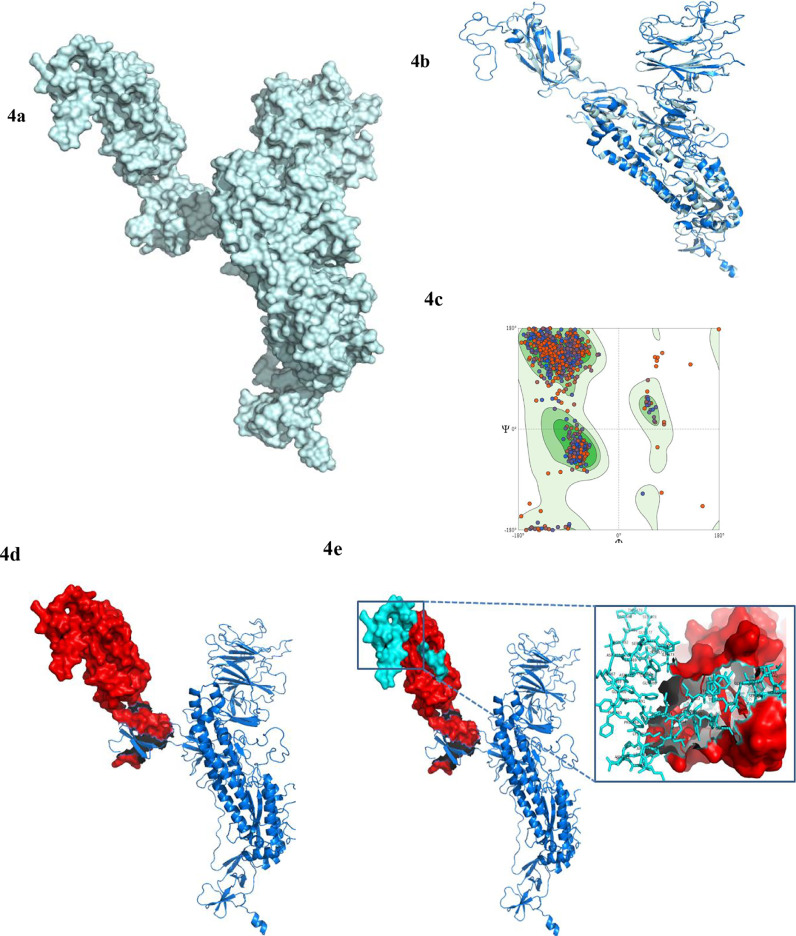
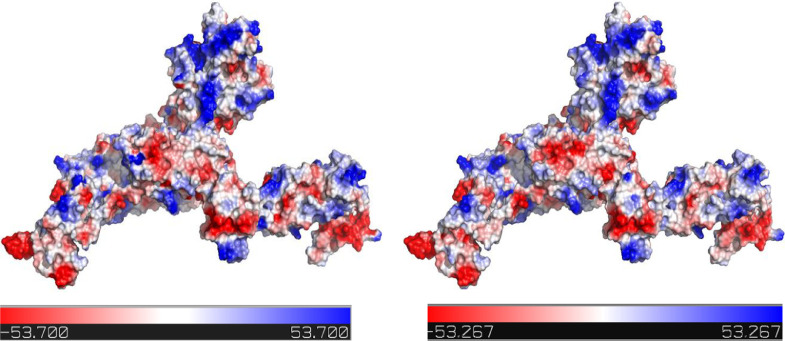
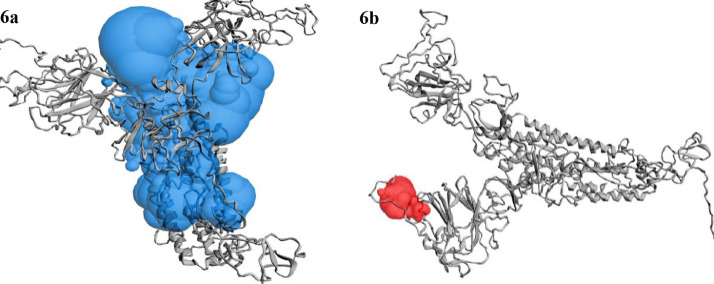
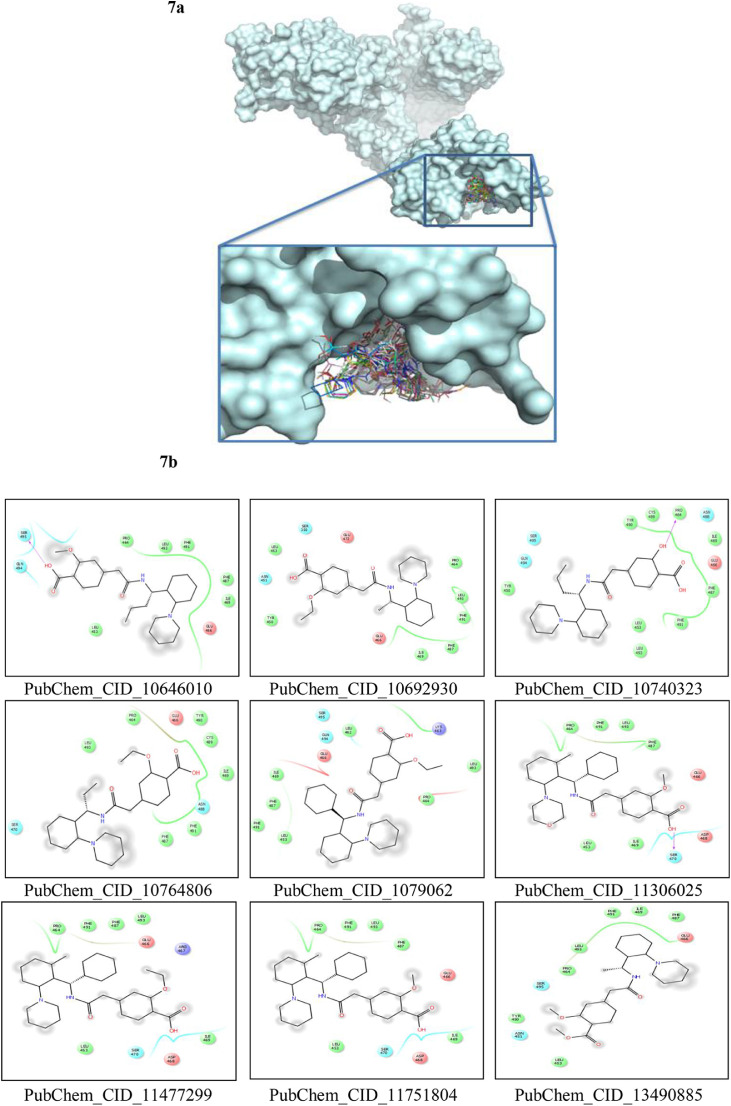
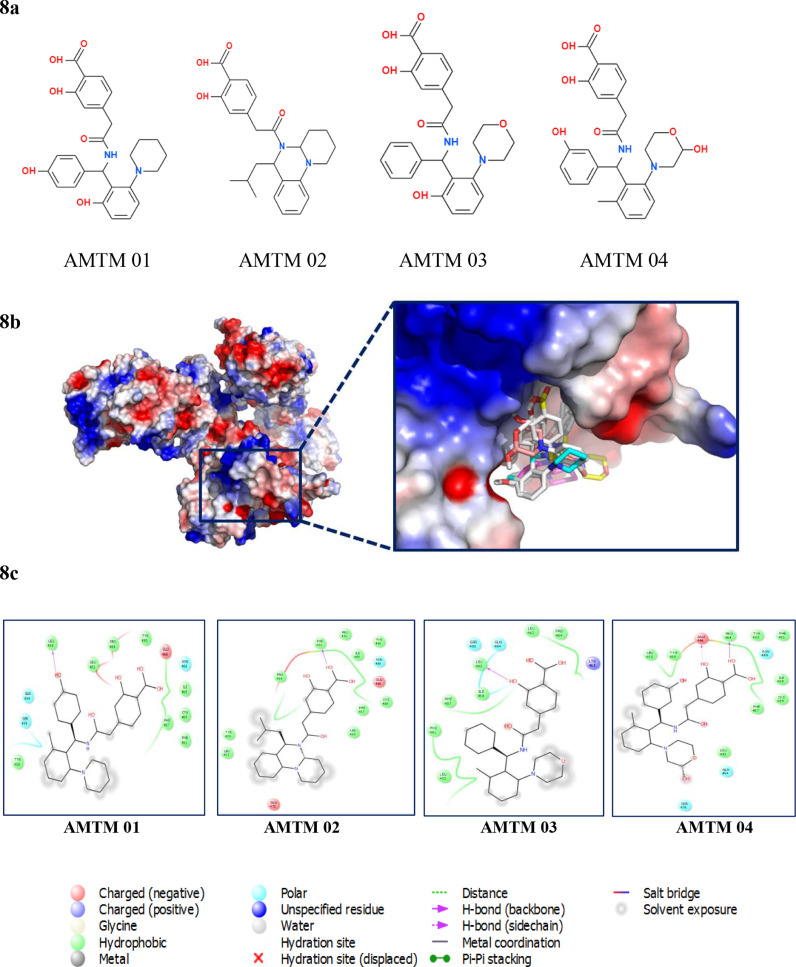
To date, the global COVID-19 pandemic has been associated with 11.8 million cases and over 545481 deaths. In this study, we have employed virtual screening approaches and selected 415 lead-like compounds from 103 million chemical substances, based on the existing drugs, from PubChem databases as potential candidates for the S protein-mediated viral attachment inhibition. Thereafter, based on drug-likeness and Lipinski's rules, 44 lead-like compounds were docked within the active side pocket of the viral-host attachment site of the S protein. Corresponding ligand properties and absorption, distribution, metabolism, excretion, and toxicity (ADMET) profile were measured. Furthermore, four novel inhibitors were designed and assessed computationally for efficacy. Comparative analysis showed the screened compounds in this study maintain better results than the proposed mother compounds, VE607 and SSAA09E2. The four designed novel lead compounds possessed more fascinating output without deviating from any of Lipinski's rules. They also showed higher bioavailability and the drug-likeness score was 0.56 and 1.81 compared with VE607 and SSAA09E2, respectively. All the screened compounds and novel compounds showed promising ADMET properties. Among them, the compound AMTM-02 was the best candidate, with a docking score of -7.5 kcal/mol. Furthermore, the binding study was verified by molecular dynamics simulation over 100 ns by assessing the stability of the complex. The proposed screened compounds and the novel compounds may give some breakthroughs for the development of a therapeutic drug to treat SARS-CoV-2 proficiently in vitro and in vivo.
截至目前,全球 COVID-19 大流行已导致 1180 万例病例和超过 545481 人死亡。在这项研究中,我们采用虚拟筛选方法,从 PubChem 数据库中现有的药物出发,选择了 1.03 亿种化学物质中的 415 种类药性化合物,作为 S 蛋白介导的病毒附着抑制的潜在候选物。此后,根据类药性和 Lipinski 规则,将 44 种类药性化合物对接至 S 蛋白病毒宿主附着位点的活性侧袋内。测量了相应的配体性质和吸收、分布、代谢、排泄和毒性(ADMET)特征。此外,还设计并计算了四种新型抑制剂的功效。对比分析表明,本研究中筛选的化合物比建议的母体化合物 VE607 和 SSAA09E2 保持更好的结果。这四种设计的新型先导化合物具有更吸引人的输出,没有偏离任何 Lipinski 规则。它们的生物利用度也更高,类药性评分分别为 0.56 和 1.81,而 VE607 和 SSAA09E2 的类药性评分分别为 0.56 和 1.81。所有筛选的化合物和新型化合物均表现出良好的 ADMET 特性。其中,化合物 AMTM-02 是最佳候选物,对接得分为-7.5 kcal/mol。此外,通过评估复合物的稳定性,通过超过 100 ns 的分子动力学模拟对结合研究进行了验证。所提出的筛选化合物和新型化合物可能为开发治疗 SARS-CoV-2 的治疗药物提供一些突破,从而在体外和体内有效地治疗 SARS-CoV-2。