Department of Biostatistics, Harvard T.H. Chan School of Public Health, Boston, MA, USA.
BMC Bioinformatics. 2020 Sep 29;21(1):424. doi: 10.1186/s12859-020-03711-2.
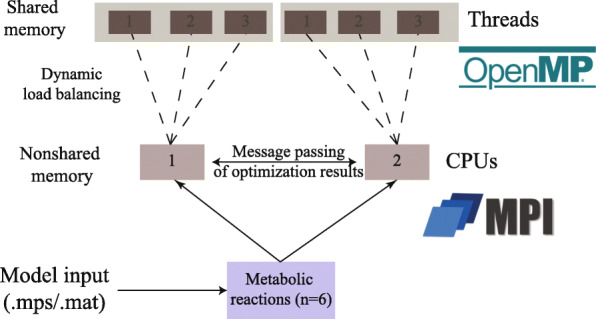
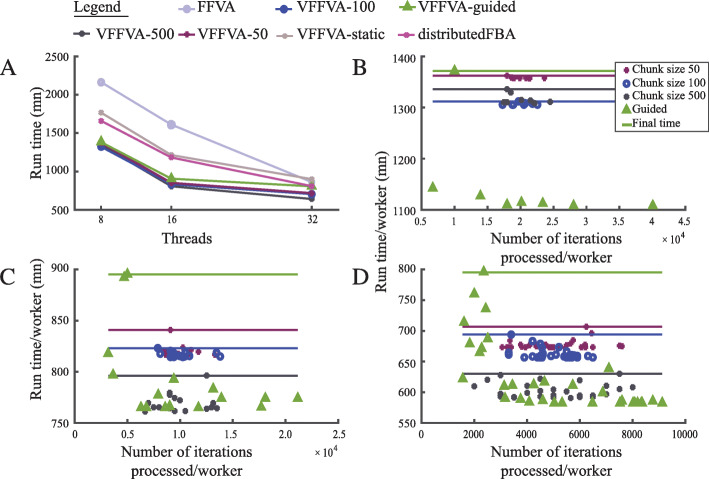
Genome-scale metabolic models are increasingly employed to predict the phenotype of various biological systems pertaining to healthcare and bioengineering. To characterize the full metabolic spectrum of such systems, Fast Flux Variability Analysis (FFVA) is commonly used in parallel with static load balancing. This approach assigns to each core an equal number of biochemical reactions without consideration of their solution complexity.
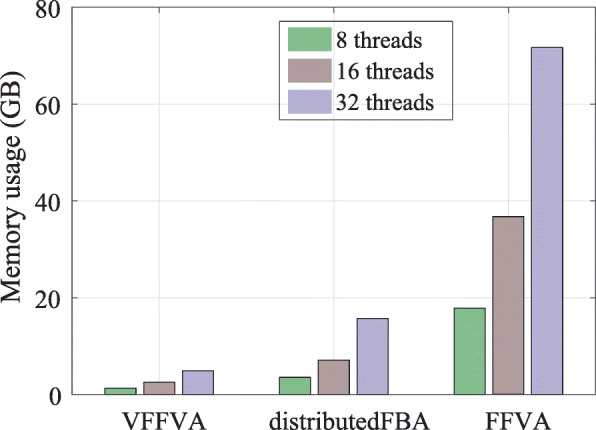
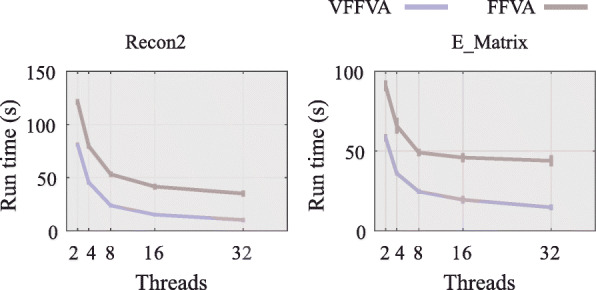
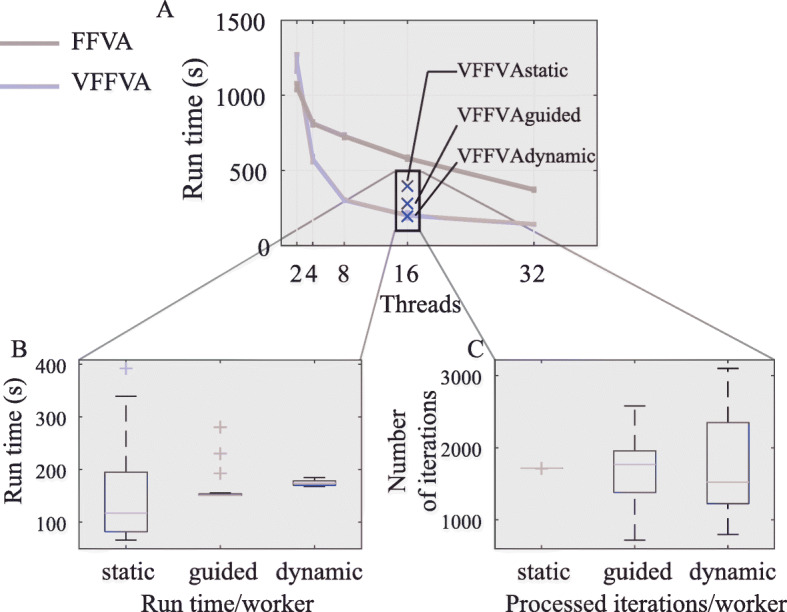
Here, we present Very Fast Flux Variability Analysis (VFFVA) as a parallel implementation that dynamically balances the computation load between the cores in runtime which guarantees equal convergence time between them. VFFVA allowed to gain a threefold speedup factor with coupled models and up to 100 with ill-conditioned models along with a 14-fold decrease in memory usage.
VFFVA exploits the parallel capabilities of modern machines to enable biological insights through optimizing systems biology modeling. VFFVA is available in C, MATLAB, and Python at https://github.com/marouenbg/VFFVA .
基因组规模的代谢模型越来越多地被用于预测与医疗保健和生物工程相关的各种生物系统的表型。为了描述这些系统的完整代谢谱,通常在并行使用快速通量变异性分析 (FFVA) 的同时进行静态负载平衡。该方法为每个核分配数量相等的生化反应,而不考虑其解决方案的复杂性。
在这里,我们提出了非常快速通量变异性分析 (VFFVA) 作为一种并行实现,它可以在运行时在核之间动态平衡计算负载,从而保证它们之间的收敛时间相等。VFFVA 允许在耦合模型中获得三倍的加速因子,在病态模型中最高可达 100 倍,同时内存使用量减少 14 倍。
VFFVA 利用现代机器的并行能力,通过优化系统生物学建模来实现生物学见解。VFFVA 可在 C、MATLAB 和 Python 中使用,网址为 https://github.com/marouenbg/VFFVA 。