Computational Molecular Evolution Group, Heidelberg Institute for Theoretical Studies, Heidelberg, Germany.
Institute for Theoretical Informatics, Karlsruhe Institute of Technology, Karlsruhe, Germany.
Mol Ecol Resour. 2021 Jan;21(1):340-349. doi: 10.1111/1755-0998.13255. Epub 2020 Oct 9.
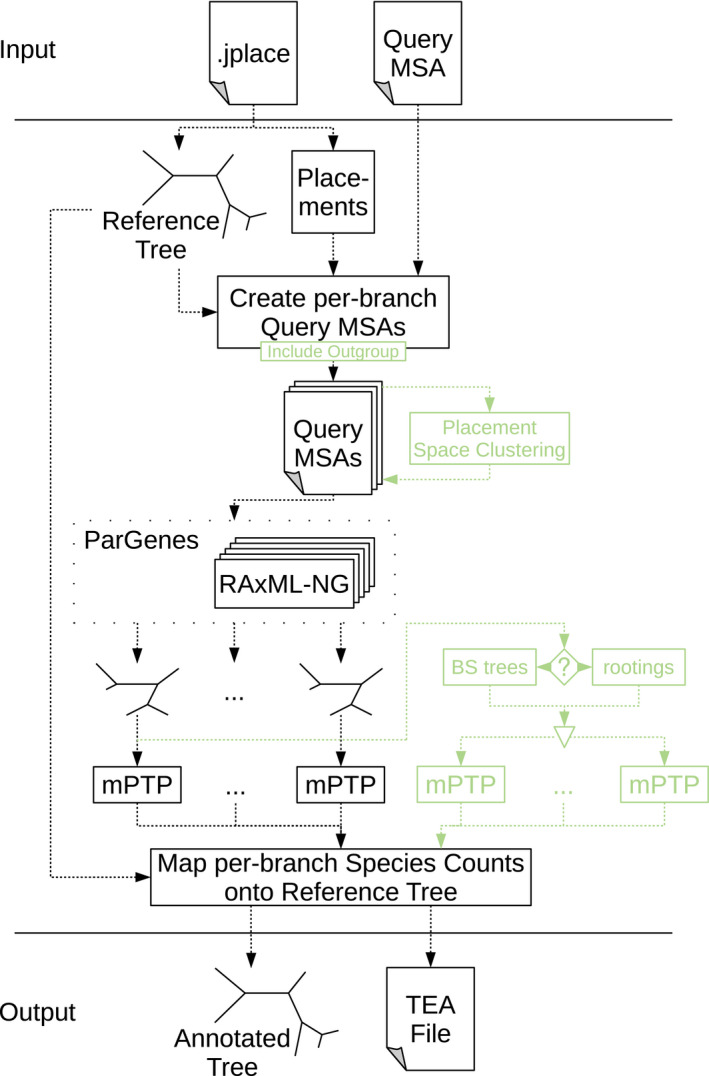
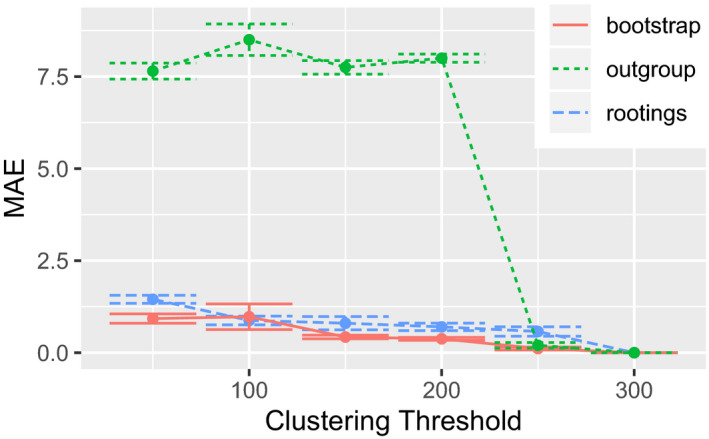
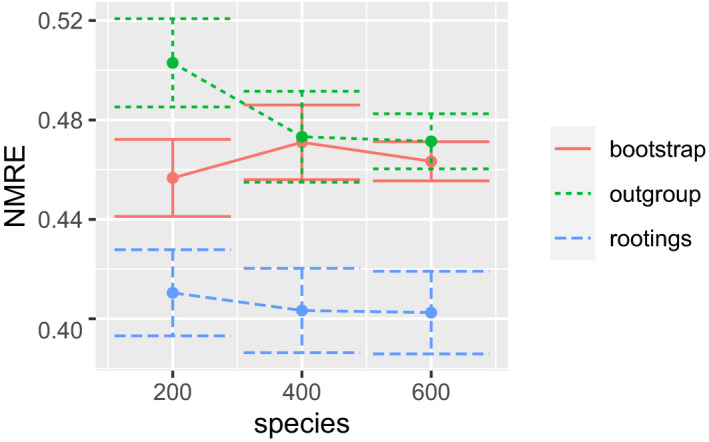
Microbial ecology research is currently driven by the continuously decreasing cost of DNA sequencing and the improving accuracy of data analysis methods. One such analysis method is phylogenetic placement, which establishes the phylogenetic identity of the anonymous environmental sequences in a sample by means of a given phylogenetic reference tree. However, assessing the diversity of a sample remains challenging, as traditional methods do not scale well with the increasing data volumes and/or do not leverage the phylogenetic placement information. Here, we present scrapp, a highly parallel and scalable tool that uses a molecular species delimitation algorithm to quantify the diversity distribution over the reference phylogeny for a given phylogenetic placement of the sample. scrapp employs a novel approach to cluster phylogenetic placements, called placement space clustering, to efficiently perform dimensionality reduction, so as to scale on large data volumes. Furthermore, it uses the phylogeny-aware molecular species delimitation method mPTP to quantify diversity. We evaluated scrapp using both, simulated and empirical data sets. We use simulated data to verify our approach. Tests on an empirical data set show that scrapp-derived metrics can classify samples by their diversity-correlated features equally well or better than existing, commonly used approaches. scrapp is available at https://github.com/pbdas/scrapp.
微生物生态学研究目前受到 DNA 测序成本不断降低和数据分析方法准确性不断提高的推动。其中一种分析方法是系统发育定位,它通过给定的系统发育参考树来确定样本中匿名环境序列的系统发育身份。然而,评估样本的多样性仍然具有挑战性,因为传统方法无法很好地扩展到不断增加的数据量,或者无法利用系统发育定位信息。在这里,我们介绍了 scrapp,这是一种高度并行和可扩展的工具,它使用分子物种划分算法来量化给定样本系统发育定位的参考系统发育上的多样性分布。scrapp 采用了一种称为放置空间聚类的新方法来对聚类进行聚类,以有效地进行降维,从而适应大数据量。此外,它还使用了基于亲缘关系的分子物种划分方法 mPTP 来量化多样性。我们使用模拟数据集和经验数据集来评估 scrapp。我们使用模拟数据来验证我们的方法。对经验数据集的测试表明,scrapp 衍生的指标可以根据其与多样性相关的特征同等或更好地对样本进行分类,而不是现有的常用方法。scrapp 可在 https://github.com/pbdas/scrapp 上获得。