Department of Life Sciences, Imperial College London, Ascot, Berks, United Kingdom.
Institute of Cell Biology and Genetic Engineering, NAS of Ukraine, Kyiv, Ukraine.
Genome Biol Evol. 2020 Sep 1;12(9):1636-1645. doi: 10.1093/gbe/evaa141.
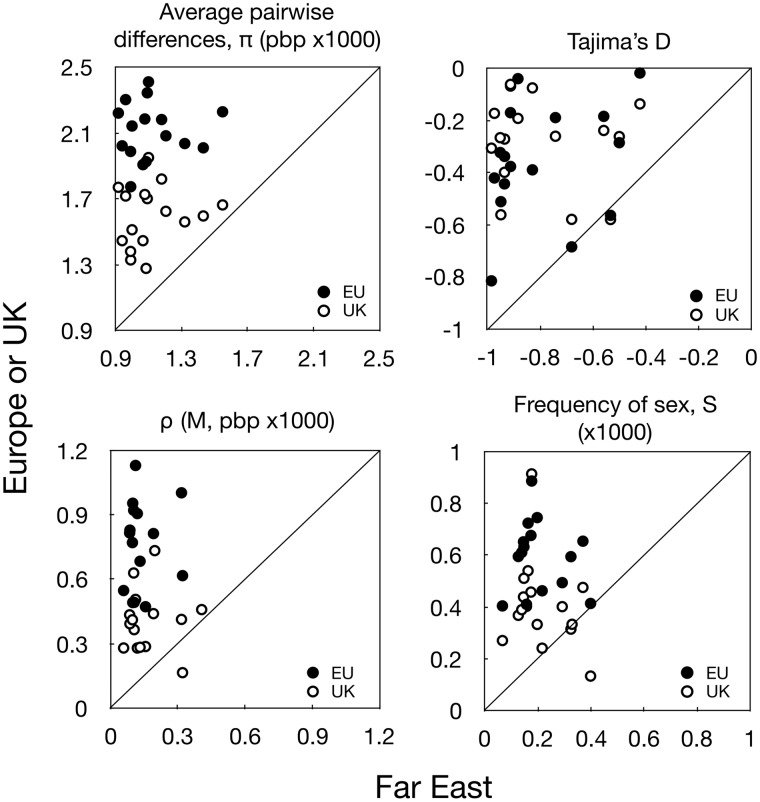
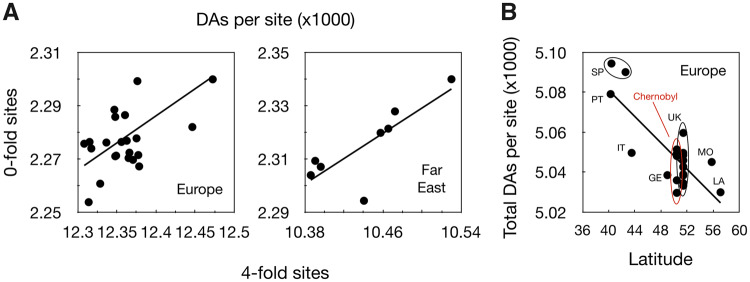
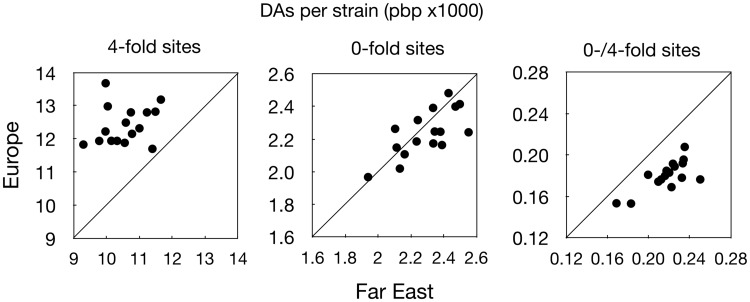
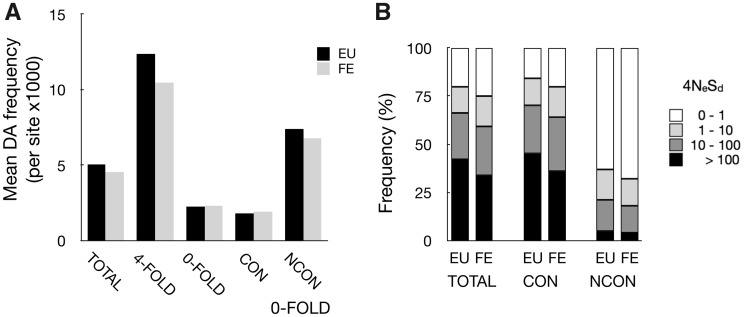
This study uses population genomic data to estimate demographic and selection parameters in two sister lineages of the wild yeast Saccharomyces paradoxus and compare their evolution. We first estimate nucleotide and recombinational diversities in each of the two lineages to infer their population size and frequency of sex and then analyze the rate of mutation accumulation since divergence from their inferred common ancestor to estimate the generation time and efficacy of selection. We find that one of the lineages has significantly higher silent nucleotide diversity and lower linkage disequilibrium, indicating a larger population with more frequent sexual generations. The same lineage also shows shorter generation time and higher efficacy of purifying selection, the latter consistent with the finding of larger population size and more frequent sex. Similar analyses are also performed on the ancestries of individual strains within lineages and we find significant differences between strains implying variation in rates of mitotic cell divisions. Our sample includes some strains originating in the Chernobyl nuclear-accident exclusion zone, which has been subjected to high levels of radiation for nearly 30 years now. We find no evidence, however, for increased rates of mutation. Finally, there is a positive correlation between rates of mutation accumulation and length of growing period, as measured by latitude of the place of origin of strains. Our study illustrates the power of genomic analyses in estimating population and life history parameters and testing predictions based on population genetic theory.
本研究利用群体基因组数据估计了野生酵母 Saccharomyces paradoxus 的两个姊妹谱系的人口统计学和选择参数,并比较了它们的进化情况。我们首先估计了两个谱系中每个谱系的核苷酸和重组多样性,以推断它们的种群大小和性别的频率,然后分析了自与推断的共同祖先分歧以来的突变积累率,以估计世代时间和选择的有效性。我们发现,其中一个谱系的沉默核苷酸多样性显著较高,连锁不平衡较低,表明种群较大,有更多的有性世代。同一谱系的世代时间也较短,纯化选择的效率较高,这与种群较大和性较频繁的发现一致。我们还对谱系内个体菌株的祖先进行了类似的分析,发现菌株之间存在显著差异,这意味着有丝分裂细胞分裂率的变化。我们的样本包括一些起源于切尔诺贝利核事故隔离区的菌株,该地区近 30 年来一直受到高水平辐射的影响。然而,我们没有发现突变率增加的证据。最后,突变积累率与菌株起源地纬度所测的生长周期长度之间存在正相关关系。我们的研究说明了基因组分析在估计种群和生活史参数以及根据群体遗传理论检验预测方面的强大功能。