Department of Ecology and Evolutionary Biology, University of Arizona, Tucson, AZ, USA.
Department of Biology, Stanford University, Stanford, CA, USA.
Philos Trans R Soc Lond B Biol Sci. 2020 Nov 23;375(1812):20190575. doi: 10.1098/rstb.2019.0575. Epub 2020 Oct 5.
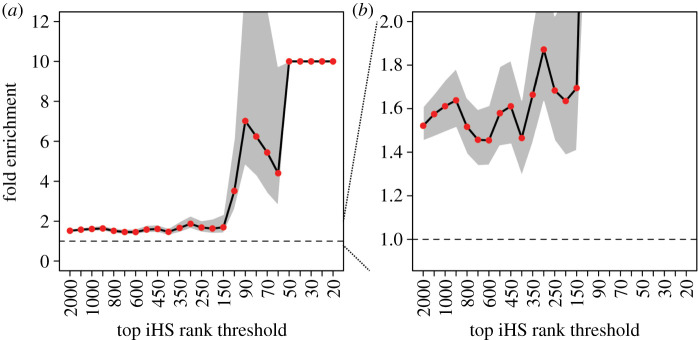
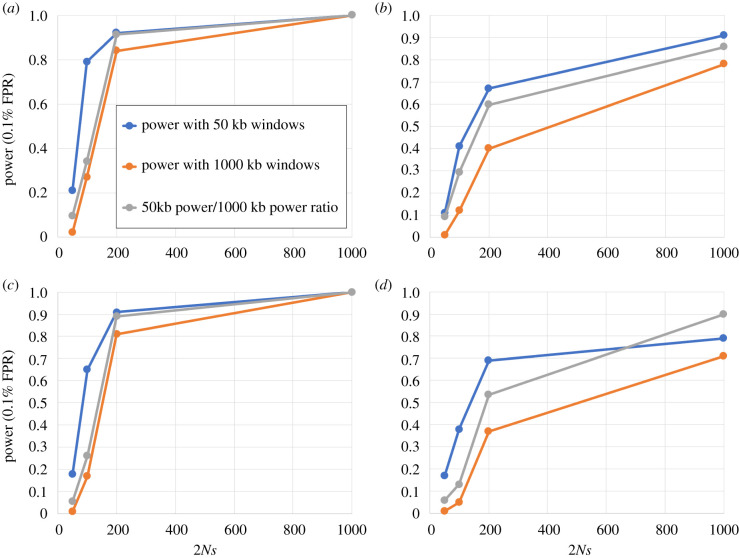
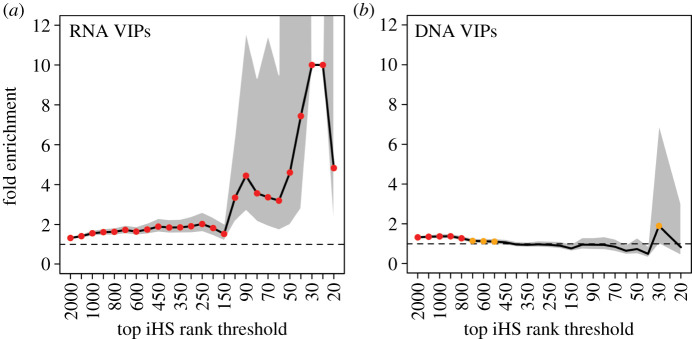
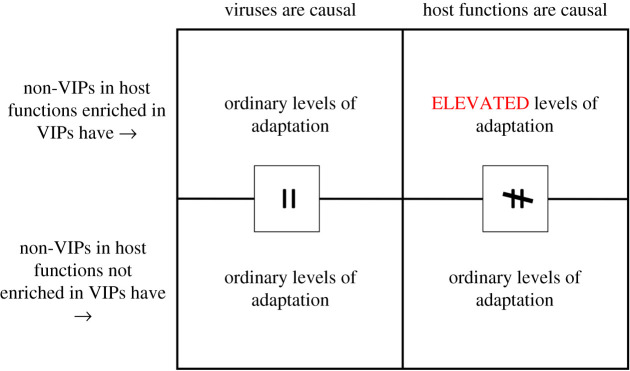
Over the course of the last several million years of evolution, humans probably have been plagued by hundreds or perhaps thousands of epidemics. Little is known about such ancient epidemics and a deep evolutionary perspective on current pathogenic threats is lacking. The study of past epidemics has typically been limited in temporal scope to recorded history, and in physical scope to pathogens that left sufficient DNA behind, such as during the Great Plague. Host genomes, however, offer an indirect way to detect ancient epidemics beyond the current temporal and physical limits. Arms races with pathogens have shaped the genomes of the hosts by driving a large number of adaptations at many genes, and these signals can be used to detect and further characterize ancient epidemics. Here, we detect the genomic footprints left by ancient viral epidemics that took place in the past approximately 50 000 years in the 26 human populations represented in the 1000 Genomes Project. By using the enrichment in signals of adaptation at approximately 4500 host loci that interact with specific types of viruses, we provide evidence that RNA viruses have driven a particularly large number of adaptive events across diverse human populations. These results suggest that different types of viruses may have exerted different selective pressures during human evolution. Knowledge of these past selective pressures will provide a deeper evolutionary perspective on current pathogenic threats. This article is part of the theme issue 'Insights into health and disease from ancient biomolecules'.
在过去的几百万年的进化过程中,人类可能遭受了数百次甚至数千次的流行病。关于这些古代流行病知之甚少,对当前致病威胁也缺乏深入的进化视角。对过去流行病的研究通常在时间范围上仅限于有记录的历史,在物理范围上仅限于留下足够 DNA 的病原体,例如大瘟疫期间。然而,宿主基因组提供了一种间接的方法,可以在当前的时间和物理限制之外检测古代流行病。与病原体的军备竞赛通过驱动许多基因的大量适应,塑造了宿主的基因组,这些信号可用于检测和进一步描述古代流行病。在这里,我们检测了在过去大约 50000 年内发生在 1000 基因组计划中代表的 26 个人类群体中的古代病毒流行病留下的基因组足迹。通过利用与特定类型病毒相互作用的大约 4500 个宿主基因座的适应性信号富集,我们提供了证据表明 RNA 病毒在不同的人类群体中引发了大量的适应性事件。这些结果表明,不同类型的病毒在人类进化过程中可能施加了不同的选择压力。了解这些过去的选择压力将为当前的致病威胁提供更深入的进化视角。本文是主题为“从古代生物分子看健康与疾病”的特刊的一部分。