Department of Cardiology, The Third People's Hospital of Shenzhen, Shenzhen 518114, China.
Department of General Surgery, Zengcheng District People's Hospital of Guangzhou, Guangzhou 511300, China.
Biomed Res Int. 2020 Sep 19;2020:8021208. doi: 10.1155/2020/8021208. eCollection 2020.
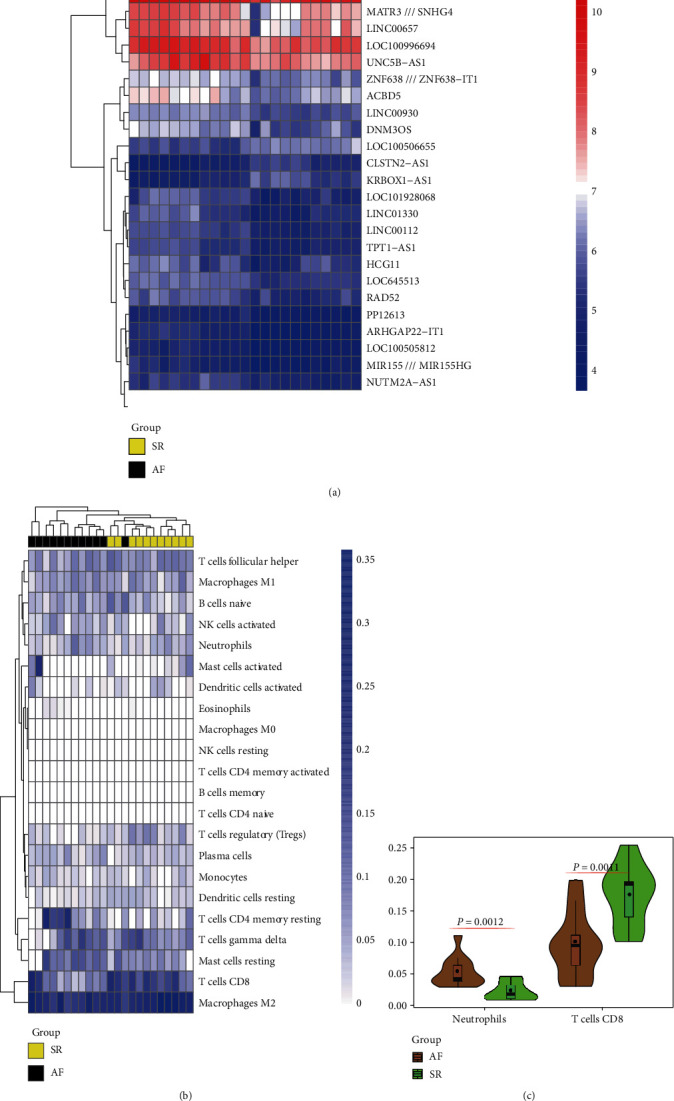
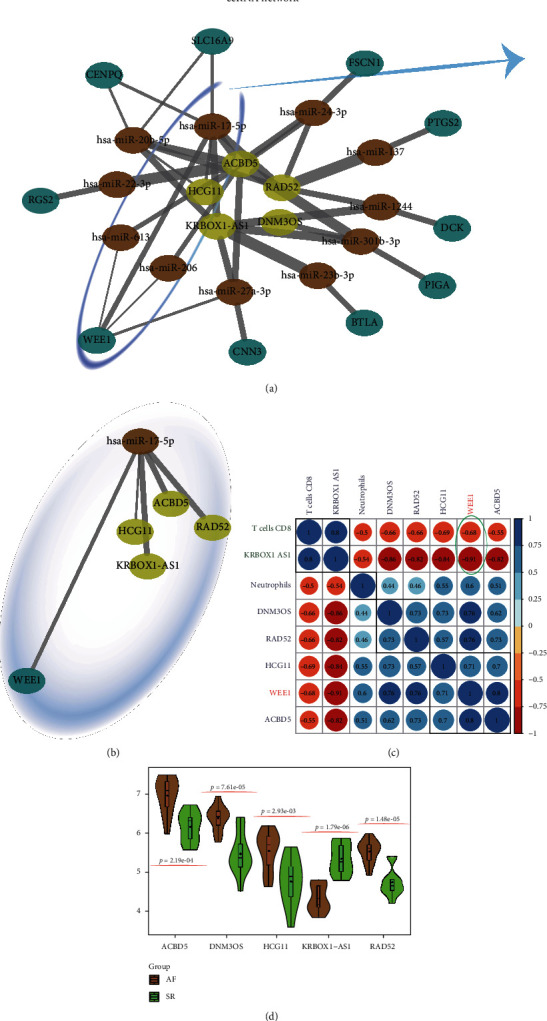
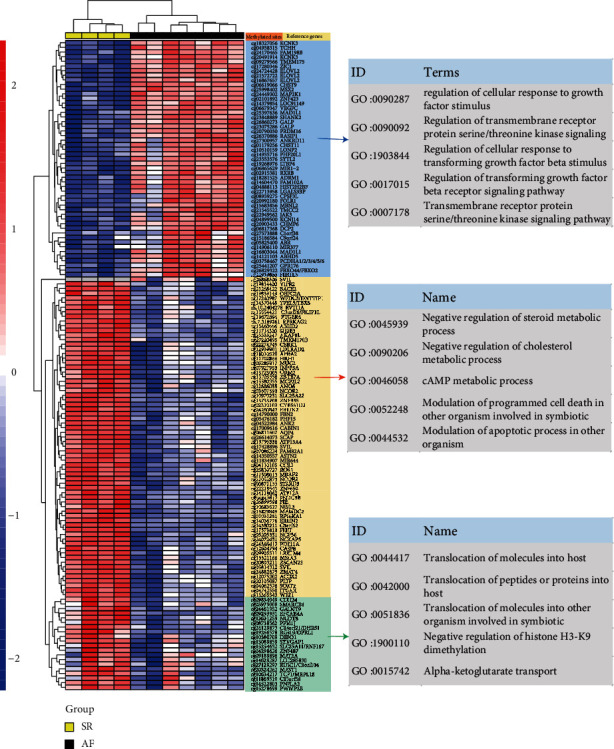
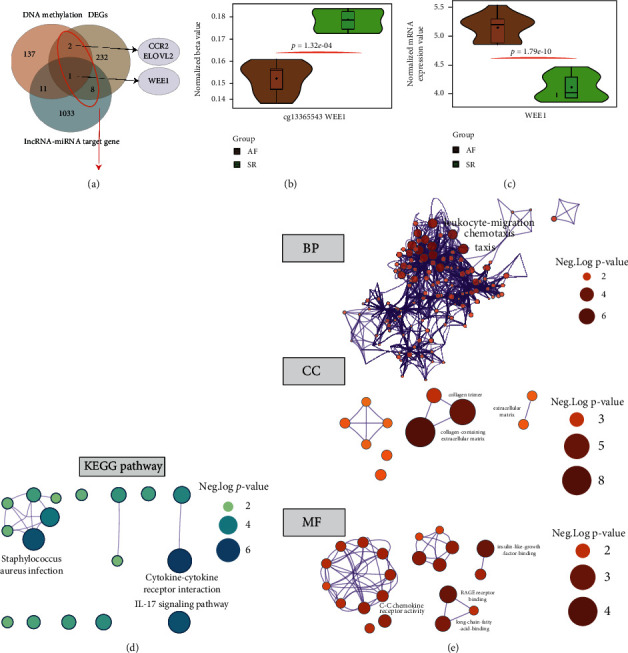
This study is aimed at identifying potential molecular mechanisms and candidate biomarkers in the left atrial regions for the diagnosis and treatment of valvular atrial fibrillation (VAF). Multibioinformatics methods, including linear models for microarray analysis (LIMMA), an SVA algorithm, CIBERSORT immune infiltration, and DNA methylation analysis, were employed. In addition, the protein-protein interaction (PPI) network, Gene Ontology (GO), and molecular pathways of differentially expressed genes (DEGs) or differential methylation regions were constructed. In all, compared with the normal rhythm group, 243 different mRNAs (29 downregulated and 214 upregulated) and 26 different lncRNAs (3 downregulated and 23 upregulated) were detected in the left atrium (LA) of atrial fibrillation (AF) patients, and the neutrophil and CD8 T cell were infiltrated. Additionally, 199 different methylation sites (107 downregulated and 92 upregulated) were also identified based on DNA methylation analysis. After integration, ELOVL2, CCR2, and WEE1 were detected for differentially methylated and differentially transcribed genes. Among them, WEE1 was also a core gene identified by the competing endogenous RNA (ceRNA) network that included WEE1-KRBOX1-AS1-hsa-miR-17-5p, in VAF left atrial tissue. We combined the DNA methylation and transcriptional expression differential analysis and found that WEE1 (cg13365543) may well be a candidate gene regulated by DNA methylation modification. Moreover, KRBOX1-AS1 and WEE1 can compete endogenously and may mediate myocardial tissue infiltration into CD8 T cells and participate in the AF process.
本研究旨在鉴定左心房区域中用于诊断和治疗瓣膜性心房颤动(VAF)的潜在分子机制和候选生物标志物。采用了多组学方法,包括微阵列分析的线性模型(LIMMA)、SVA 算法、CIBERSORT 免疫浸润和 DNA 甲基化分析。此外,还构建了蛋白质-蛋白质相互作用(PPI)网络、基因本体论(GO)和差异表达基因(DEGs)或差异甲基化区域的分子途径。总之,与正常节律组相比,在心房颤动(AF)患者的左心房(LA)中检测到 243 个不同的 mRNA(29 个下调和 214 个上调)和 26 个不同的 lncRNA(3 个下调和 23 个上调),并且中性粒细胞和 CD8 T 细胞浸润。此外,还基于 DNA 甲基化分析鉴定了 199 个不同的甲基化位点(107 个下调和 92 个上调)。整合后,检测到 ELOVL2、CCR2 和 WEE1 存在差异甲基化和转录基因。其中,WEE1 也是包括 WEE1-KRBOX1-AS1-hsa-miR-17-5p 在内的 VAF 左心房组织中 ceRNA 网络的核心基因。我们结合 DNA 甲基化和转录表达差异分析发现,WEE1(cg13365543)可能是受 DNA 甲基化修饰调节的候选基因。此外,KRBOX1-AS1 和 WEE1 可以内源性竞争,并可能介导 CD8 T 细胞浸润心肌组织,参与 AF 过程。