UC Berkeley-UCSF Graduate Program in Bioengineering, University of California San Francisco, San Francisco, CA, United States of America.
Department of Bioengineering and Therapeutic Sciences, University of California San Francisco, San Francisco, CA, United States of America.
PLoS Comput Biol. 2020 Oct 5;16(10):e1008178. doi: 10.1371/journal.pcbi.1008178. eCollection 2020 Oct.
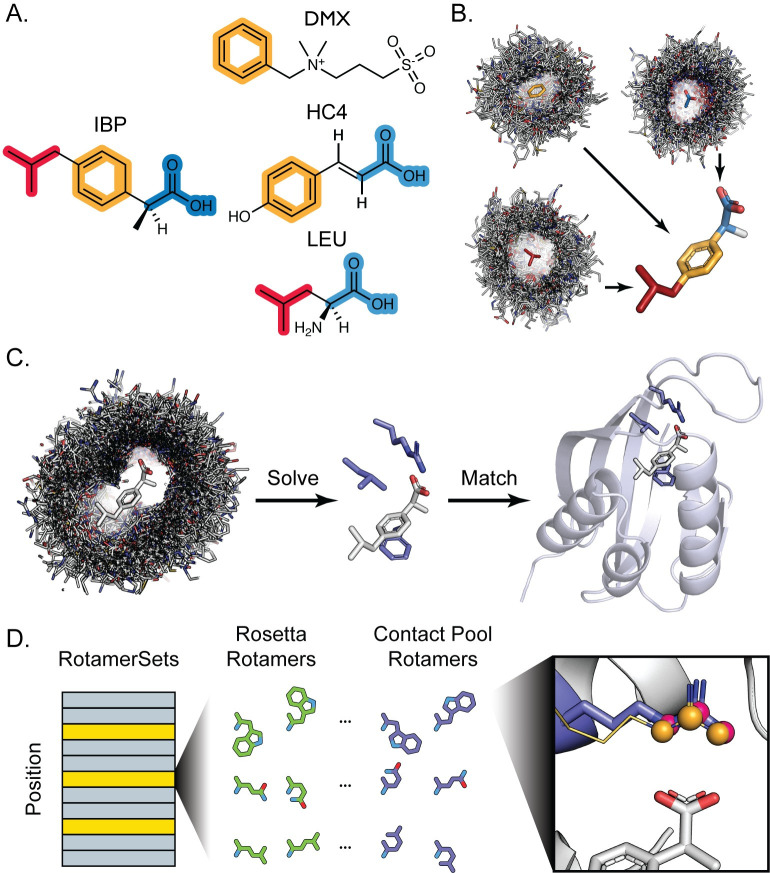
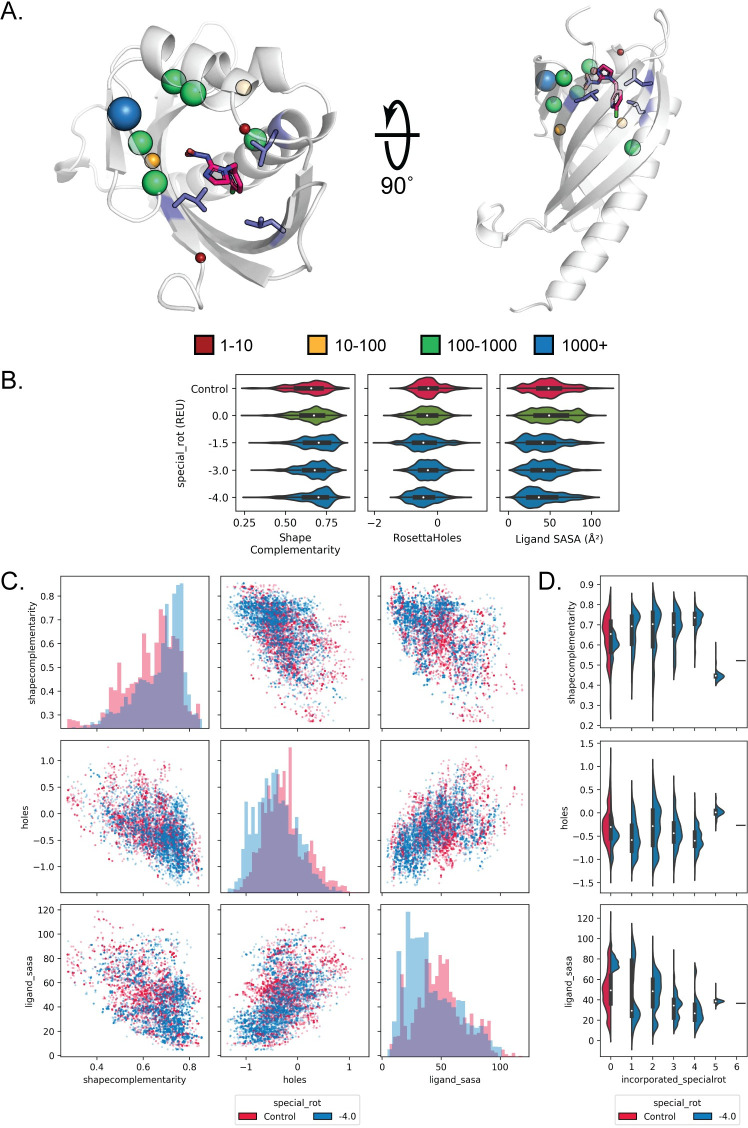
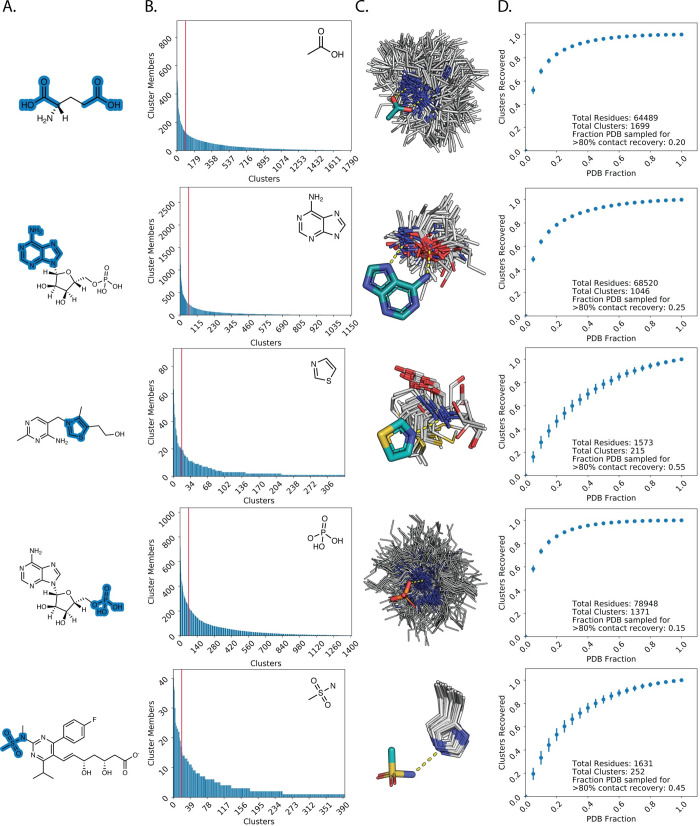
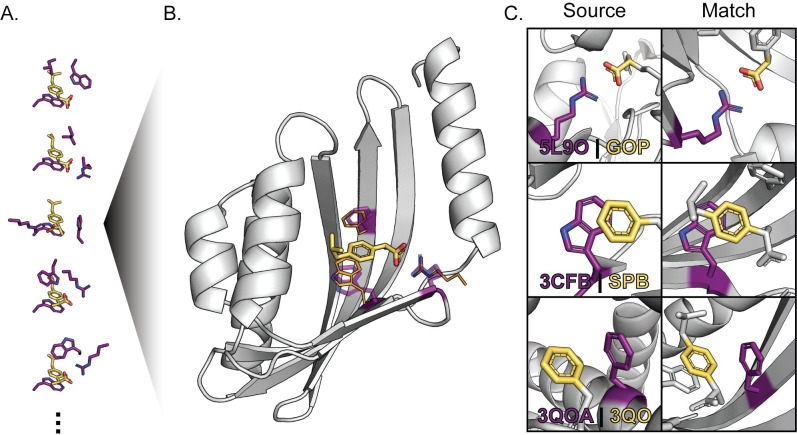
Protein binding to small molecules is fundamental to many biological processes, yet it remains challenging to predictively design this functionality de novo. Current state-of-the-art computational design methods typically rely on existing small molecule binding sites or protein scaffolds with existing shape complementarity for a target ligand. Here we introduce new methods that utilize pools of discrete contacts between protein side chains and defined small molecule ligand substructures (ligand fragments) observed in the Protein Data Bank. We use the Rosetta Molecular Modeling Suite to recombine protein side chains in these contact pools to generate hundreds of thousands of energetically favorable binding sites for a target ligand. These composite binding sites are built into existing scaffold proteins matching the intended binding site geometry with high accuracy. In addition, we apply pools of side chain rotamers interacting with the target ligand to augment Rosetta's conventional design machinery and improve key metrics known to be predictive of design success. We demonstrate that our method reliably builds diverse binding sites into different scaffold proteins for a variety of target molecules. Our generalizable de novo ligand binding site design method provides a foundation for versatile design of protein to interface previously unattainable molecules for applications in medical diagnostics and synthetic biology.
蛋白质与小分子的结合对于许多生物过程至关重要,但从头预测设计这种功能仍然具有挑战性。目前最先进的计算设计方法通常依赖于现有小分子结合位点或具有现有形状互补性的蛋白质支架,以适应目标配体。在这里,我们引入了新的方法,利用蛋白质侧链与定义明确的小分子配体亚结构(配体片段)之间离散接触的池,这些接触在蛋白质数据库中观察到。我们使用 Rosetta 分子建模套件来重组这些接触池中蛋白质侧链,为目标配体生成数十万能量有利的结合位点。这些复合结合位点构建到与预期结合位点几何形状高度匹配的现有支架蛋白中。此外,我们应用与目标配体相互作用的侧链旋转异构体池来增强 Rosetta 的常规设计机制,并提高已知可预测设计成功的关键指标。我们证明我们的方法能够可靠地将不同的结合位点构建到不同的支架蛋白中,适用于各种目标分子。我们可推广的从头开始配体结合位点设计方法为蛋白质界面设计提供了基础,以前无法实现的分子可以应用于医学诊断和合成生物学。