National Institute of Pharmaceutical Education and Research, Kolkata 70054, India.
Institute for Bioinformatics and Medical Informatics, University of Tübingen, Sand 14, 72076 Tübingen, Germany.
Molecules. 2020 Oct 10;25(20):4604. doi: 10.3390/molecules25204604.
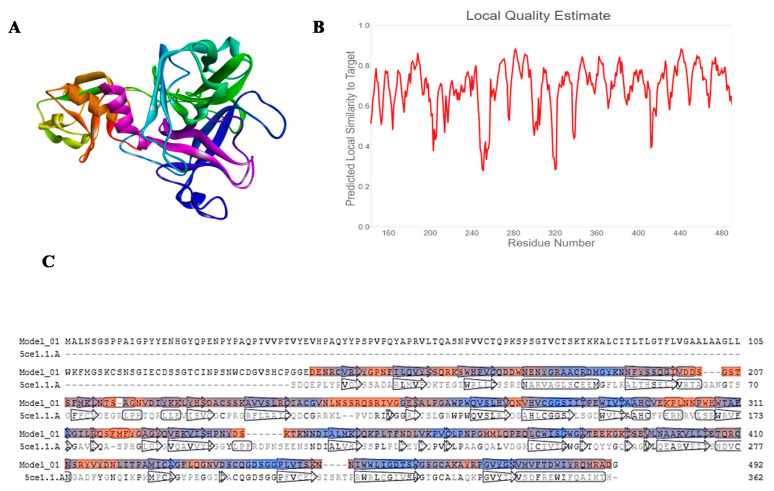
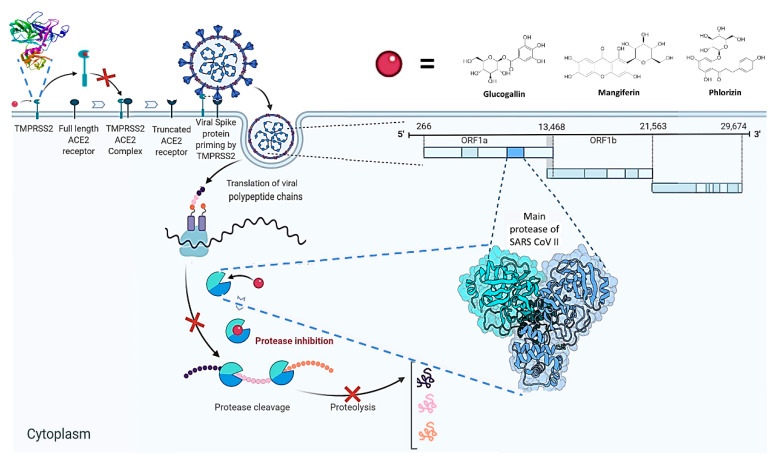
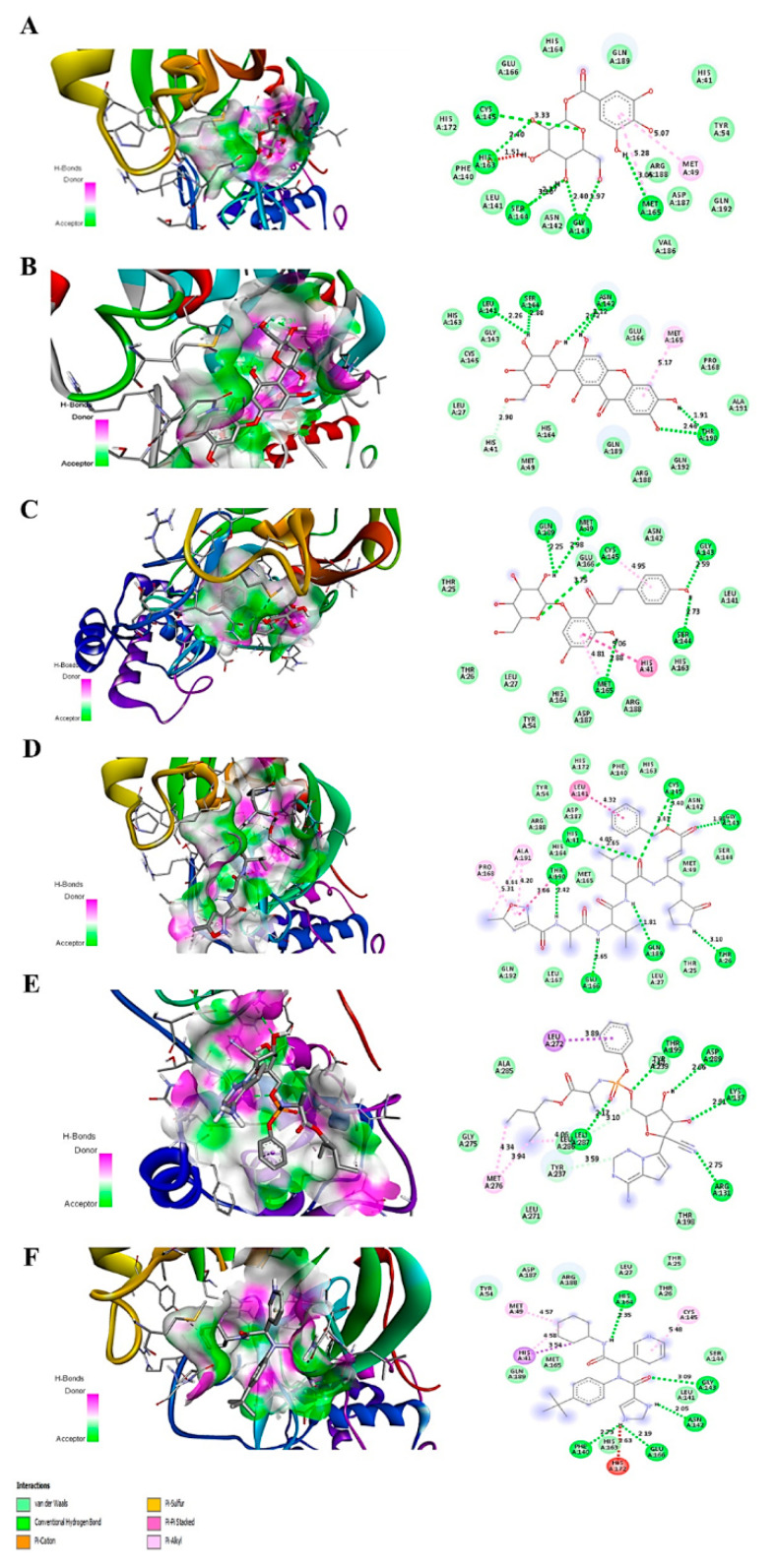
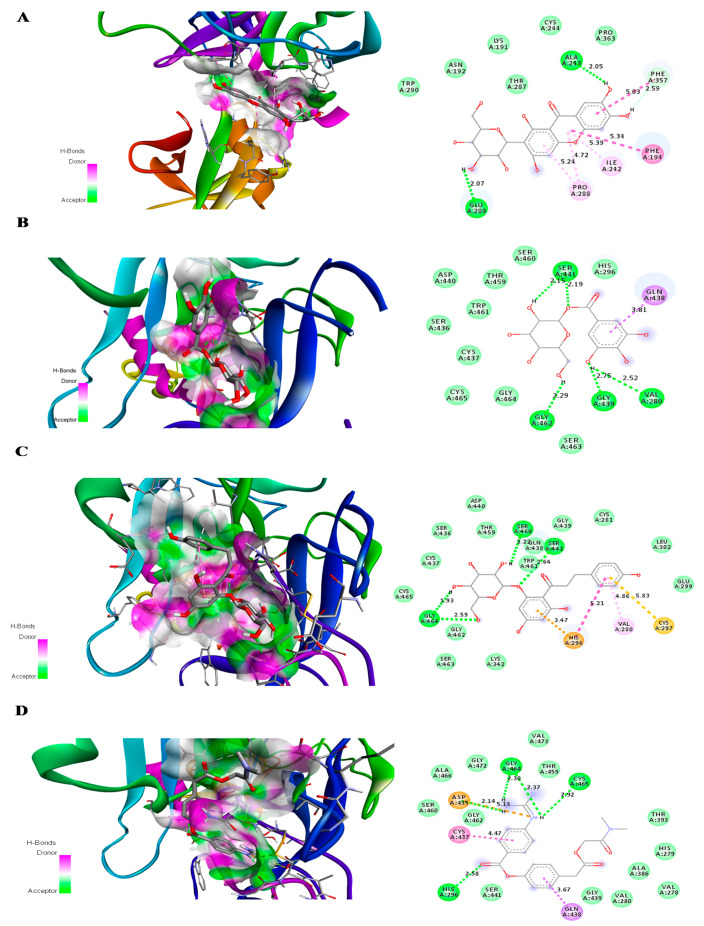
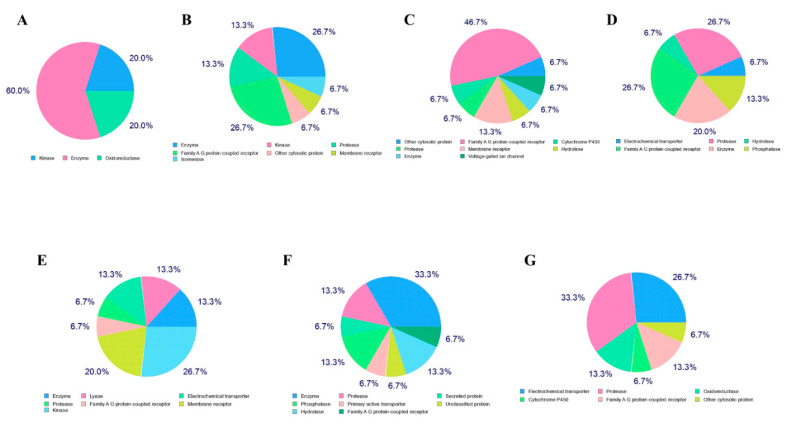
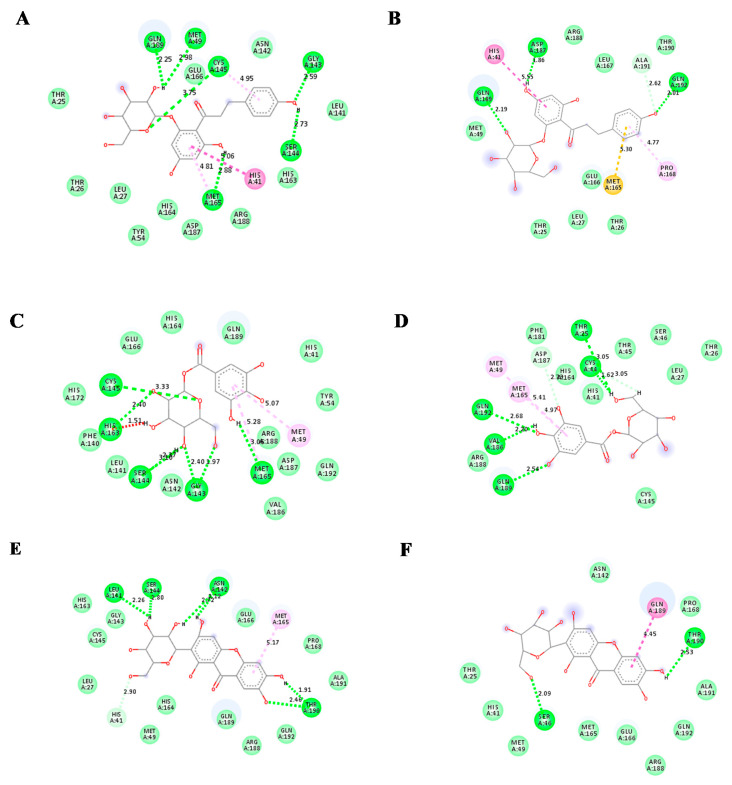
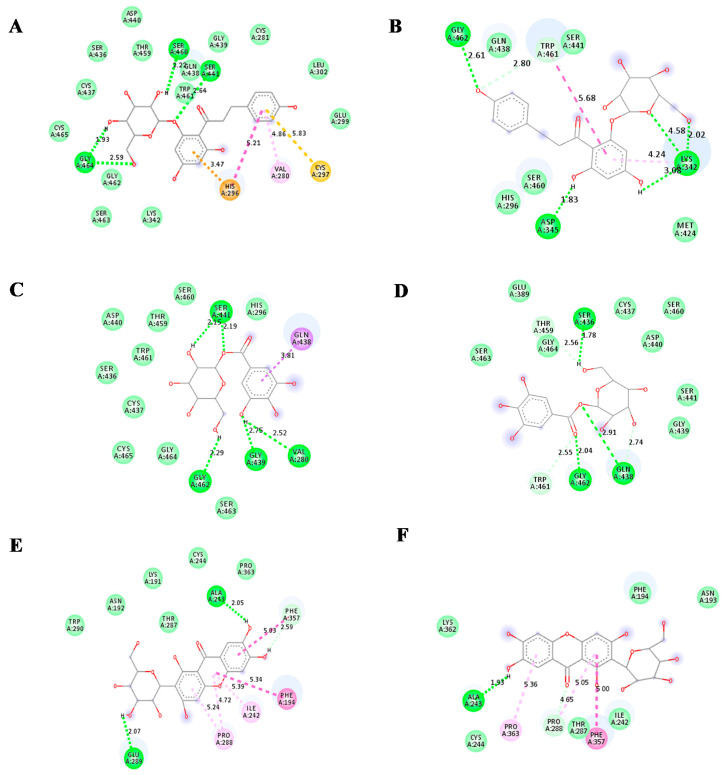
The current pandemic, caused by SARS-CoV-2 virus, is a severe challenge for human health and the world economy. There is an urgent need for development of drugs that can manage this pandemic, as it has already infected 19 million people and led to the death of around 711,277 people worldwide. At this time, in-silico studies are providing lots of preliminary data about potential drugs, which can be a great help in further in-vitro and in-vivo studies. Here, we have selected three polyphenolic compounds, mangiferin, glucogallin, and phlorizin. These compounds are isolated from different natural sources but share structural similarities and have been reported for their antiviral activity. The objective of this study is to analyze and predict the anti-protease activity of these compounds on SARS-CoV-2main protease (Mpro) and TMPRSS2 protein. Both the viral protein and the host protein play an important role in the viral life cycle, such as post-translational modification and viral spike protein priming. This study has been performed by molecular docking of the compounds using PyRx with AutoDock Vina on the two aforementioned targets chosen for this study, i.e., SARS-CoV-2 Mpro and TMPRSS2. The compounds showed good binding affinity and are further analyzed by (Molecular dynamic) MD and Molecular Mechanics Poisson-Boltzmann Surface Area MM-PBSA study. The MD-simulation study has predicted that these natural compounds will have a great impact on the stabilization of the binding cavity of the Mpro of SARS-CoV-2. The predicted pharmacokinetic parameters also show that these compounds are expected to have good solubility and absorption properties. Further predictions for these compounds also showed no involvement in drug-drug interaction and no toxicity.
当前由 SARS-CoV-2 病毒引起的大流行对人类健康和世界经济构成了严重挑战。迫切需要开发能够应对这种大流行的药物,因为它已经感染了 1900 万人,并导致全球约 711277 人死亡。此时,计算机模拟研究为潜在药物提供了大量初步数据,这对进一步的体外和体内研究将有很大帮助。在这里,我们选择了三种多酚类化合物,芒果苷、葡萄糖醛酸和根皮苷。这些化合物分别从不同的天然来源中分离出来,但具有结构相似性,并已被报道具有抗病毒活性。本研究旨在分析和预测这些化合物对 SARS-CoV-2 主要蛋白酶(Mpro)和 TMPRSS2 蛋白的抗蛋白酶活性。病毒蛋白和宿主蛋白在病毒生命周期中都起着重要作用,如翻译后修饰和病毒刺突蛋白的启动。这项研究是通过使用 PyRx 与 AutoDock Vina 对这两种选择的研究靶点,即 SARS-CoV-2 Mpro 和 TMPRSS2 上的化合物进行分子对接来完成的。这些化合物显示出良好的结合亲和力,并通过(分子动态)MD 和分子力学泊松-玻尔兹曼表面积 MM-PBSA 研究进一步分析。MD 模拟研究预测,这些天然化合物将对 SARS-CoV-2 Mpro 的结合腔的稳定性产生重大影响。预测的药代动力学参数还表明,这些化合物有望具有良好的溶解度和吸收特性。对这些化合物的进一步预测还表明它们不会参与药物相互作用,也没有毒性。