Department of Molecular Endocrinology, National Research Institute for Child Health and Development, Tokyo, Japan.
Laboratory of Health Nutrition, Department of Applied Biological Chemistry, Graduate School of Agricultural and Life Sciences, The University of Tokyo, Tokyo, Japan.
Sci Rep. 2020 Oct 15;10(1):17375. doi: 10.1038/s41598-020-74405-1.
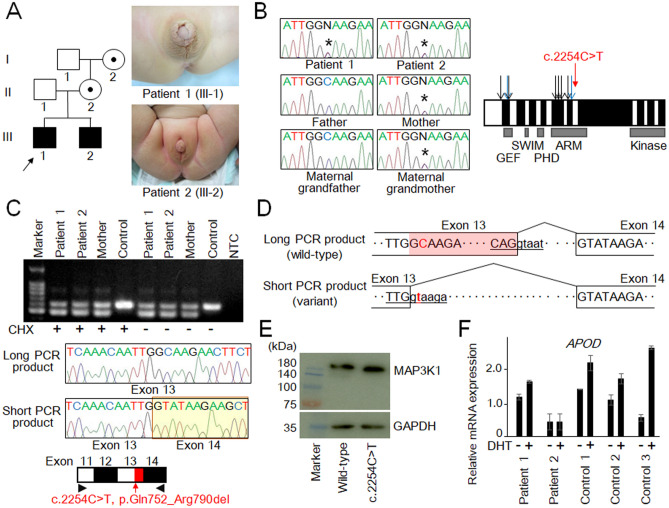
Although splicing errors due to single nucleotide variants represent a common cause of monogenic disorders, only a few variants have been shown to create new splice sites in exons. Here, we report an MAP3K1 splice variant identified in two siblings with 46,XY disorder of sex development. The patients carried a maternally derived c.2254C>T variant. The variant was initially recognized as a nonsense substitution leading to nonsense-mediated mRNA decay (p.Gln752Ter); however, RT-PCR for lymphoblastoid cell lines showed that this variant created a new splice donor site and caused 39 amino acid deletion (p.Gln752_Arg790del). All transcripts from the variant allele appeared to undergo altered splicing. The two patients exhibited undermasculinized genitalia with and without hypergonadotropism. Testosterone enanthate injections and dihydrotestosterone ointment applications yielded only slight increase in their penile length. Dihydrotestosterone-induced APOD transactivation was less significant in patients' genital skin fibroblasts compared with that in control samples. This study provides an example of nonsense-associated altered splicing, in which a highly potent exonic splice site was created. Furthermore, our data, in conjunction with the previous data indicating the association between MAP3K1 and androgen receptor signaling, imply that the combination of testicular dysgenesis and androgen insensitivity may be a unique phenotype of MAP3K1 abnormalities.
虽然由于单核苷酸变异导致的剪接错误是单基因疾病的常见原因,但仅有少数变异已被证明能在外显子中创建新的剪接位点。在这里,我们报道了在两个性发育障碍 46,XY 患者的兄弟姐妹中发现的 MAP3K1 剪接变异体。患者携带母源性 c.2254C>T 变异。该变异最初被识别为导致无义介导的 mRNA 降解的无义突变(p.Gln752Ter);然而,对淋巴母细胞系的 RT-PCR 显示,该变异创建了一个新的剪接供体位点,并导致 39 个氨基酸缺失(p.Gln752_Arg790del)。来自变异等位基因的所有转录本似乎都经历了改变的剪接。这两个患者表现为生殖器未充分男性化,伴有或不伴有高促性腺激素血症。睾酮戊酸酯注射和二氢睾酮软膏应用仅使他们的阴茎长度略有增加。与对照样本相比,患者生殖器皮肤成纤维细胞中二氢睾酮诱导的 APOD 反式激活作用较弱。本研究提供了一个无义相关剪接改变的例子,其中创建了一个高度有效的外显子剪接位点。此外,我们的数据结合先前表明 MAP3K1 与雄激素受体信号之间关联的数据,表明睾丸发育不良和雄激素不敏感的组合可能是 MAP3K1 异常的独特表型。