Faculty of Agriculture, Shizuoka University, 836 Ohya, Suruga-ku, Shizuoka, 422-8529, Japan.
United Graduate School of Agricultural Science, Gifu University, 1-1 Yanagito, Gifu, 501-1193, Japan.
Sci Rep. 2020 Oct 15;10(1):17480. doi: 10.1038/s41598-020-74623-7.
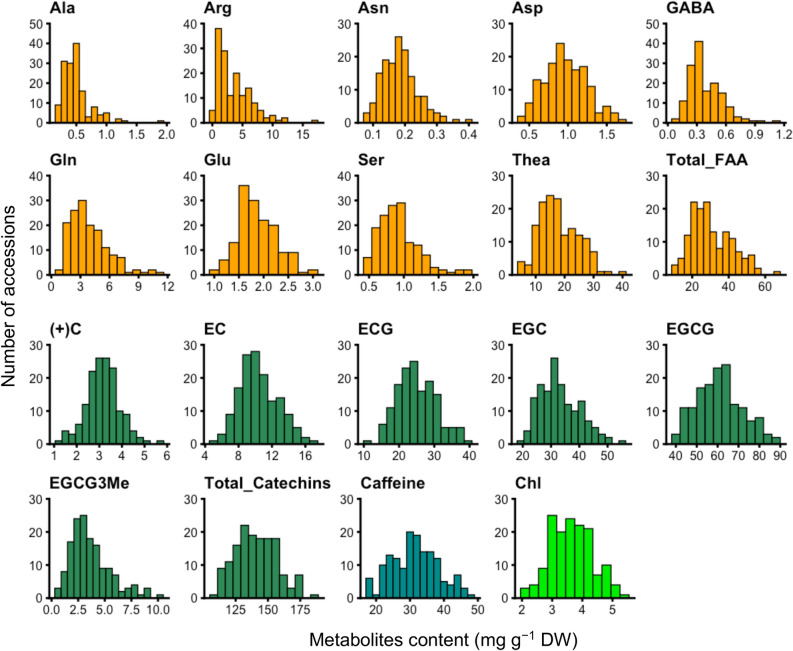
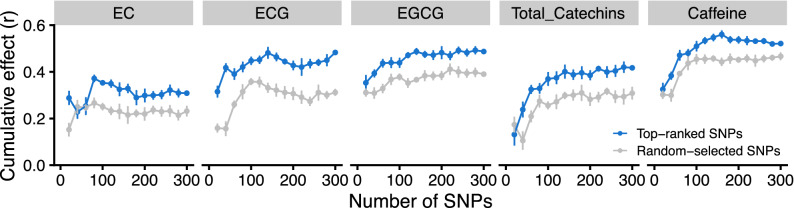
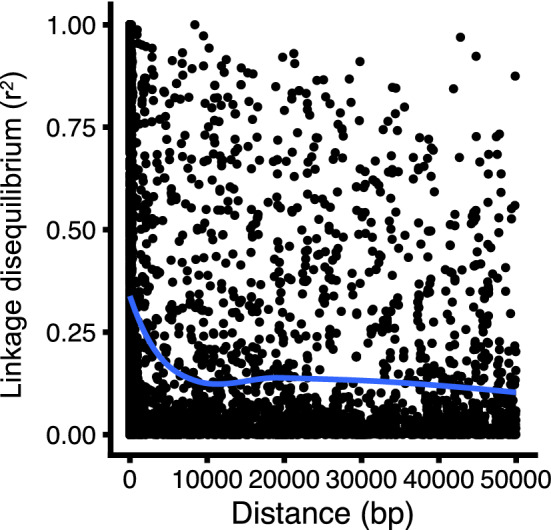
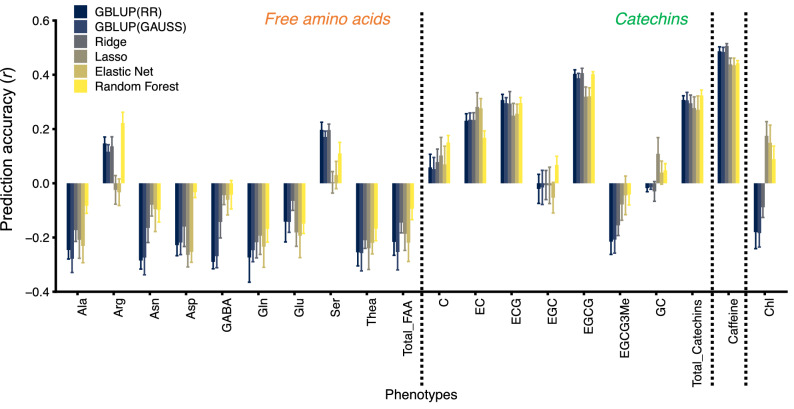
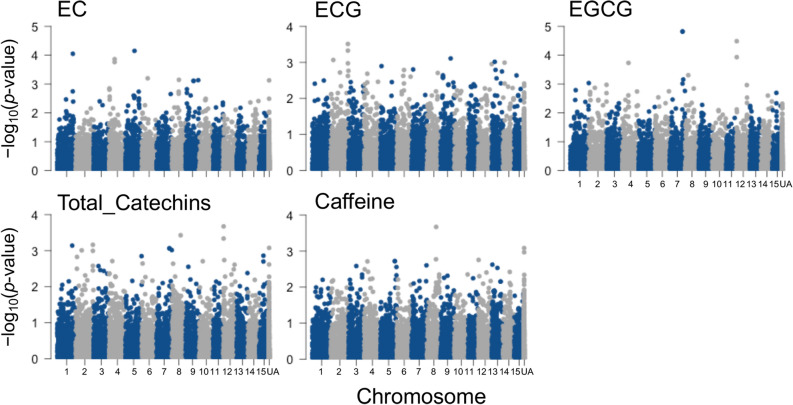
Effectively using genomic information greatly accelerates conventional breeding and applying it to long-lived crops promotes the conversion to genomic breeding. Because tea plants are bred using conventional methods, we evaluated the potential of genomic predictions (GPs) and genome-wide association studies (GWASs) for the genetic breeding of tea quality-related metabolites using genome-wide single nucleotide polymorphisms (SNPs) detected from restriction site-associated DNA sequencing of 150 tea accessions. The present GP, based on genome-wide SNPs, and six models produced moderate prediction accuracy values (r) for the levels of most catechins, represented by ( -)-epigallocatechin gallate (r = 0.32-0.41) and caffeine (r = 0.44-0.51), but low r values for free amino acids and chlorophylls. Integrated analysis of GWAS and GP detected potential candidate genes for each metabolite using 80-160 top-ranked SNPs that resulted in the maximum cumulative prediction value. Applying GPs and GWASs to tea accession traits will contribute to genomics-assisted tea breeding.
有效利用基因组信息极大地加速了常规育种,将其应用于长寿命作物促进了向基因组育种的转变。由于茶树是通过常规方法进行培育的,我们评估了基因组预测(GP)和全基因组关联研究(GWAS)在利用从 150 个茶树品种的限制性位点相关 DNA 测序中检测到的全基因组单核苷酸多态性(SNP)进行与茶叶品质相关的代谢物遗传育种方面的潜力。本研究基于全基因组 SNP,使用六种模型对大多数儿茶素(以(-)-表没食子儿茶素没食子酸酯(r=0.32-0.41)和咖啡因(r=0.44-0.51)为代表)的水平进行了中等预测准确性值(r)的预测,但对游离氨基酸和叶绿素的 r 值较低。使用产生最大累积预测值的 80-160 个排名最高的 SNP,对 GWAS 和 GP 进行综合分析,检测到每种代谢物的潜在候选基因。将 GP 和 GWASs 应用于茶树品种性状将有助于开展基于基因组学的茶树育种。