Department of Health Promotion, Mother and Child Care, Internal Medicine and Medical Specialties, University of Palermo, 90127 Palermo, Italy.
Department of Biomedicine, Neuroscience and Advanced Diagnostics (BIND), University of Palermo, 90127 Palermo, Italy.
Int J Mol Sci. 2020 Oct 15;21(20):7631. doi: 10.3390/ijms21207631.
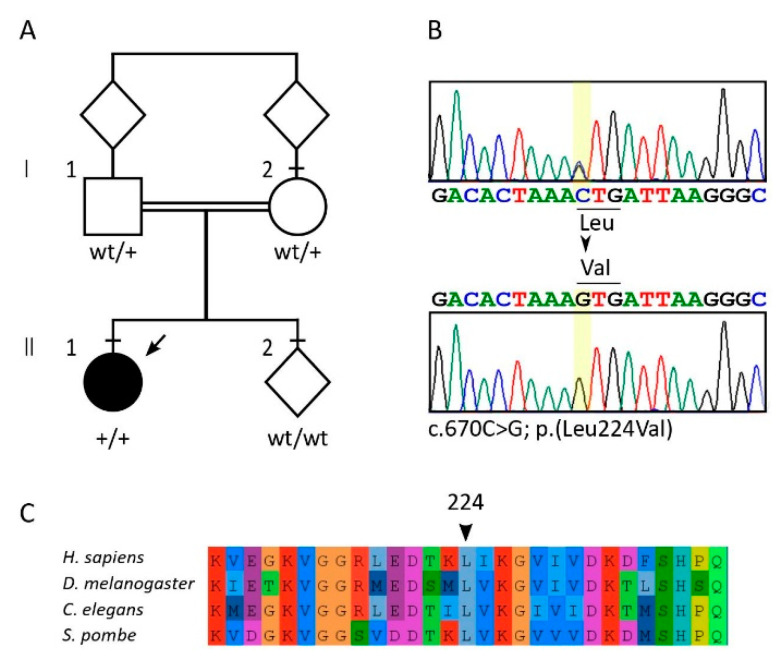
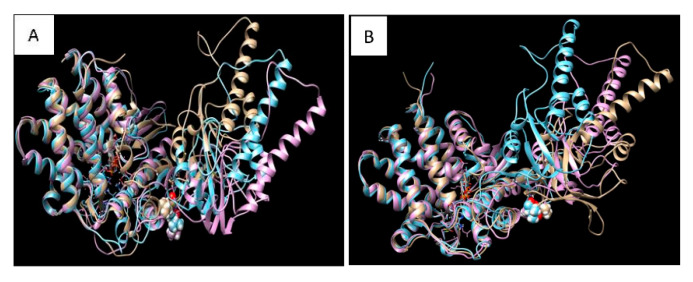
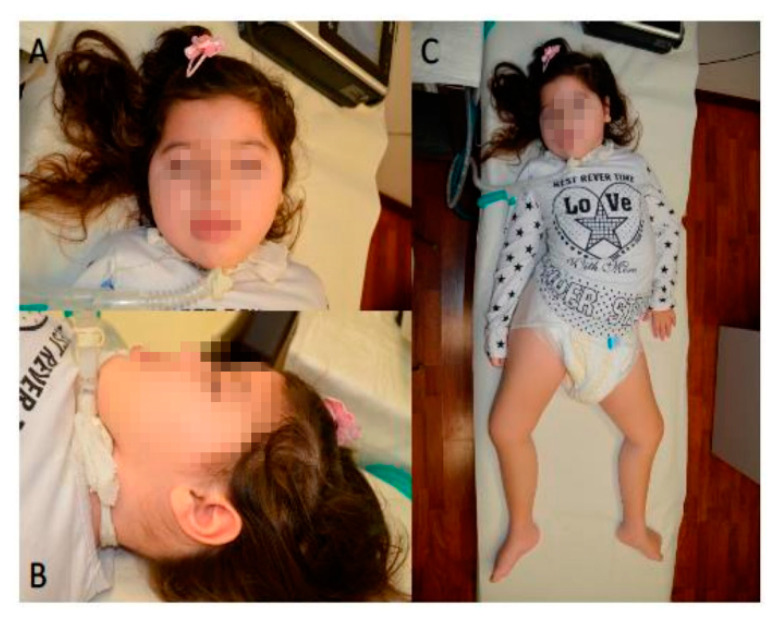
Diseases associated with acquired or genetic defects in members of the chaperoning system (CS) are increasingly found and have been collectively termed chaperonopathies. Illustrative instances of genetic chaperonopathies involve the genes for chaperonins of Groups I (e.g., Heat shock protein 60, ) and II (e.g., Chaperonin Containing T-Complex polypeptide 1, ). Examples of the former are hypomyelinating leukodystrophy 4 (HLD4 or MitCHAP60) and hereditary spastic paraplegia (SPG13). A distal sensory mutilating neuropathy has been linked to a mutation [p.(His147Arg)] in subunit 5 of the gene. Here, we describe a new possibly pathogenic variant [p.(Leu224Val)] of the same subunit but with a different phenotype. This yet undescribed disease affects a girl with early onset demyelinating neuropathy and a severe motor disability. By whole exome sequencing (WES), we identified a homozygous c.670C>G p.(Leu224Val) variant in the gene. In silico 3D-structure analysis and bioinformatics indicated that this variant could undergo abnormal conformation and could be pathogenic. We compared the patient's clinical, neurophysiological and laboratory data with those from patients carrying p.(His147Arg) in the equatorial domain. Our patient presented signs and symptoms absent in the p.(His147Arg) cases. Molecular dynamics simulation and modelling showed that the Leu224Val mutation that occurs in the CCT5 intermediate domain near the apical domain induces a conformational change in the latter. Noteworthy is the striking difference between the phenotypes putatively linked to mutations in the same CCT subunit but located in different structural domains, offering a unique opportunity for elucidating their distinctive roles in health and disease.
越来越多的疾病与伴侣蛋白系统(CS)成员中获得性或遗传性缺陷有关,这些疾病被统称为伴侣蛋白病。遗传性伴侣蛋白病的典型例子包括 I 组(例如热休克蛋白 60)和 II 组(例如伴侣蛋白含 T 复合物多肽 1)伴侣蛋白的基因。前者的例子包括低髓鞘白质营养不良 4(HLD4 或 MitCHAP60)和遗传性痉挛性截瘫(SPG13)。远端感觉性破坏性神经病与基因亚单位 5 的突变 [p.(His147Arg)]有关。在这里,我们描述了同一亚单位的另一种可能致病的变体 [p.(Leu224Val)],但表型不同。这种尚未描述的疾病影响了一名女孩,她患有早期脱髓鞘性神经病和严重的运动障碍。通过全外显子组测序(WES),我们在 基因中发现了一个纯合的 c.670C>G p.(Leu224Val)变体。计算机 3D 结构分析和生物信息学表明,这种变体可能发生异常构象,具有致病性。我们将患者的临床、神经生理学和实验室数据与携带赤道域中 p.(His147Arg)的患者进行了比较。我们的患者表现出了在 p.(His147Arg)病例中不存在的体征和症状。分子动力学模拟和建模表明,发生在 CCT5 中间域靠近顶端域的 Leu224Val 突变会导致后者构象发生变化。值得注意的是,假定与 CCT 亚单位突变相关的表型位于不同的结构域,这为阐明它们在健康和疾病中的独特作用提供了一个独特的机会。