Health Research Institute of Santiago de Compostela (IDIS), Santiago de Compostela, Spain.
Department of Hematology, SERGAS, Complexo Hospitalario Universitario de Santiago de Compostela (CHUS), Santiago, Spain.
BMC Cancer. 2020 Oct 21;20(1):1017. doi: 10.1186/s12885-020-07492-y.
Thirty to forty percent of patients with Diffuse Large B-cell Lymphoma (DLBCL) have an adverse clinical evolution. The increased understanding of DLBCL biology has shed light on the clinical evolution of this pathology, leading to the discovery of prognostic factors based on gene expression data, genomic rearrangements and mutational subgroups. Nevertheless, additional efforts are needed in order to enable survival predictions at the patient level. In this study we investigated new machine learning-based models of survival using transcriptomic and clinical data.
Gene expression profiling (GEP) of in 2 different publicly available retrospective DLBCL cohorts were analyzed. Cox regression and unsupervised clustering were performed in order to identify probes associated with overall survival on the largest cohort. Random forests were created to model survival using combinations of GEP data, COO classification and clinical information. Cross-validation was used to compare model results in the training set, and Harrel's concordance index (c-index) was used to assess model's predictability. Results were validated in an independent test set.
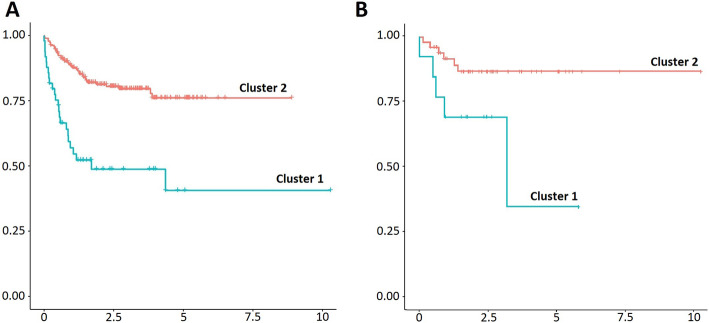
Two hundred thirty-three and sixty-four patients were included in the training and test set, respectively. Initially we derived and validated a 4-gene expression clusterization that was independently associated with lower survival in 20% of patients. This pattern included the following genes: TNFRSF9, BIRC3, BCL2L1 and G3BP2. Thereafter, we applied machine-learning models to predict survival. A set of 102 genes was highly predictive of disease outcome, outperforming available clinical information and COO classification. The final best model integrated clinical information, COO classification, 4-gene-based clusterization and the expression levels of 50 individual genes (training set c-index, 0.8404, test set c-index, 0.7942).
Our results indicate that DLBCL survival models based on the application of machine learning algorithms to gene expression and clinical data can largely outperform other important prognostic variables such as disease stage and COO. Head-to-head comparisons with other risk stratification models are needed to compare its usefulness.
30%至 40%弥漫性大 B 细胞淋巴瘤(DLBCL)患者的临床预后不良。对 DLBCL 生物学的深入了解揭示了该疾病的临床演变,并导致基于基因表达数据、基因组重排和突变亚群发现预后因素。然而,仍需要进一步努力,以便能够对患者层面的生存进行预测。在这项研究中,我们使用转录组学和临床数据研究了新的基于机器学习的生存模型。
对两个不同的公开可用的回顾性 DLBCL 队列的基因表达谱(GEP)进行了分析。在最大的队列中,进行 Cox 回归和无监督聚类以识别与总生存相关的探针。使用 GEP 数据、COO 分类和临床信息的组合创建随机森林来建立生存模型。交叉验证用于比较训练集中的模型结果,哈雷尔一致性指数(c-index)用于评估模型的可预测性。结果在独立的测试集中得到验证。
分别有 233 名和 64 名患者纳入训练集和测试集。最初,我们推导并验证了一种 4 个基因表达聚类,该聚类在 20%的患者中与较低的生存率独立相关。该模式包括以下基因:TNFRSF9、BIRC3、BCL2L1 和 G3BP2。此后,我们应用机器学习模型来预测生存。一组 102 个基因对疾病结局具有高度预测性,优于现有的临床信息和 COO 分类。最终最佳模型集成了临床信息、COO 分类、基于 4 个基因的聚类以及 50 个单个基因的表达水平(训练集 c-index,0.8404,测试集 c-index,0.7942)。
我们的结果表明,基于机器学习算法应用于基因表达和临床数据的 DLBCL 生存模型可以大大优于其他重要的预后变量,如疾病分期和 COO。需要与其他风险分层模型进行头对头比较,以比较其有用性。