Department of Psychiatry and Behavioral Neurosciences, Wayne State University School of Medicine, Detroit, MI, USA.
Department of Psychiatry, University of Michigan, Ann Arbor, MI, USA.
Sci Rep. 2020 Oct 21;10(1):17935. doi: 10.1038/s41598-020-74481-3.
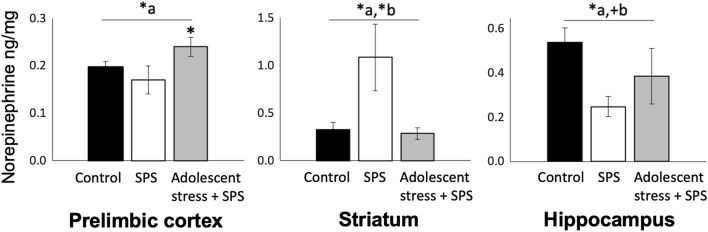
Stress in adolescence can regulate vulnerability to traumatic stress in adulthood through region-specific epigenetic activity and catecholamine levels. We hypothesized that stress in adolescence would increase adult trauma vulnerability by impairing extinction-retention, a deficit in PTSD, by (1) altering class IIa histone deacetylases (HDACs), which integrate effects of stress on gene expression, and (2) enhancing norepinephrine in brain regions regulating cognitive effects of trauma. We investigated the effects of adolescent-stress on adult vulnerability to severe stress using the single-prolonged stress (SPS) model in male rats. Rats were exposed to either (1) adolescent-stress (33-35 postnatal days) then SPS (58-60 postnatal days; n = 14), or (2) no adolescent-stress and SPS (58-60 postnatal days; n = 14), or (3) unstressed conditions (n = 8). We then measured extinction-retention, norepinephrine, HDAC4, and HDAC5. As expected, SPS exposure induced an extinction-retention deficit. Adolescent-stress prior to SPS eliminated this deficit, suggesting adolescent-stress conferred resiliency to adult severe stress. Adolescent-stress also conferred region-specific resilience to norepinephrine changes. HDAC4 and HDAC5 were down-regulated following SPS, and these changes were also modulated by adolescent-stress. Regulation of HDAC levels was consistent with the pattern of cognitive effects of SPS; only animals exposed to SPS without adolescent-stress exhibited reduced HDAC4 and HDAC5 in the prelimbic cortex, hippocampus, and striatum. Thus, HDAC regulation caused by severe stress in adulthood interacts with stress history such that seemingly conflicting reports describing effects of adolescent stress on adult PTSD vulnerability may stem in part from dynamic HDAC changes following trauma that are shaped by adolescent stress history.
青少年时期的压力会通过特定区域的表观遗传活性和儿茶酚胺水平来调节成年后对创伤性压力的易感性。我们假设,青少年时期的压力会通过以下两种方式损害 PTSD 的消退-保持,从而增加成年后创伤的易感性:(1)改变 IIa 类组蛋白去乙酰化酶(HDACs),这些酶整合了压力对基因表达的影响;(2)增强调节创伤认知效应的大脑区域中的去甲肾上腺素。我们使用雄性大鼠的单一延长压力(SPS)模型研究了青少年压力对成年后对严重压力的易感性的影响。大鼠经历以下三种情况之一:(1)青少年压力(33-35 日龄),然后是 SPS(58-60 日龄;n=14);(2)无青少年压力和 SPS(58-60 日龄;n=14);或(3)未受压力的条件(n=8)。然后,我们测量了消退-保持、去甲肾上腺素、HDAC4 和 HDAC5。正如预期的那样,SPS 暴露会导致消退-保持缺陷。SPS 之前的青少年压力消除了这种缺陷,这表明青少年压力赋予了成年后严重压力的弹性。青少年压力也赋予了去甲肾上腺素变化的特定区域的弹性。SPS 后,HDAC4 和 HDAC5 下调,这些变化也受到青少年压力的调节。HDAC 水平的调节与 SPS 的认知效应模式一致;只有未经历青少年压力的 SPS 暴露的动物在额前皮质、海马体和纹状体中表现出 HDAC4 和 HDAC5 的减少。因此,成年后严重应激引起的 HDAC 调节与应激史相互作用,使得描述青少年应激对成年 PTSD 易感性影响的看似矛盾的报告可能部分源于创伤后动态 HDAC 变化,而这些变化是由青少年应激史塑造的。