School of Biomedical Sciences, Faculty of Biological Sciences, University of Leeds, UK.
Sino-UK Joint Laboratory of Brain Function and Injury of Henan Province and Department of Physiology and Pathophysiology, Xinxiang Medical University, PR China; School of Biomedical Sciences, Faculty of Biological Sciences, University of Leeds, UK.
Redox Biol. 2020 Oct;37:101755. doi: 10.1016/j.redox.2020.101755. Epub 2020 Oct 16.
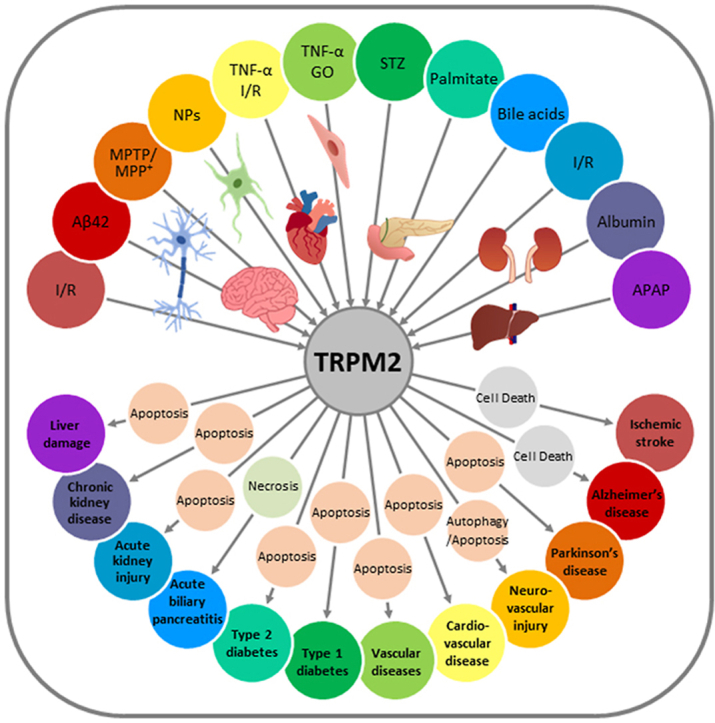
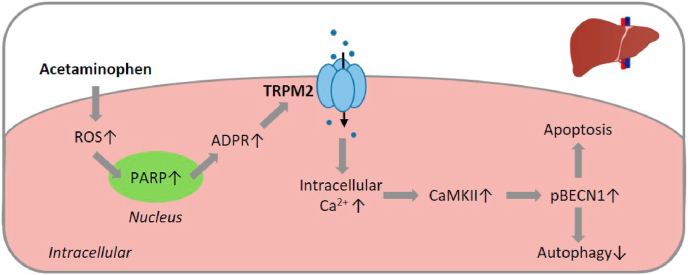
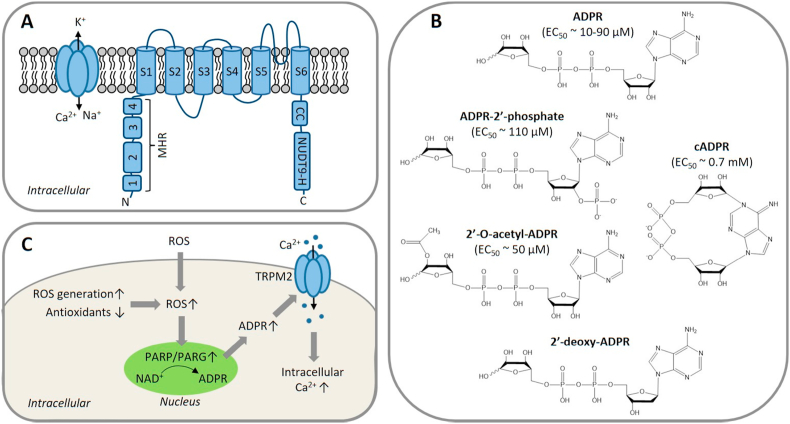
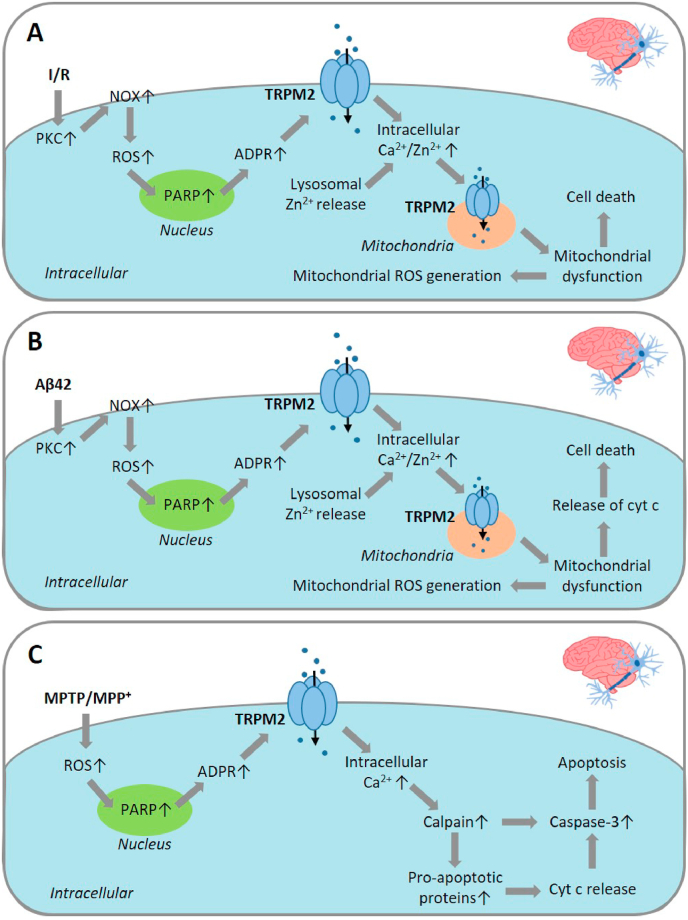
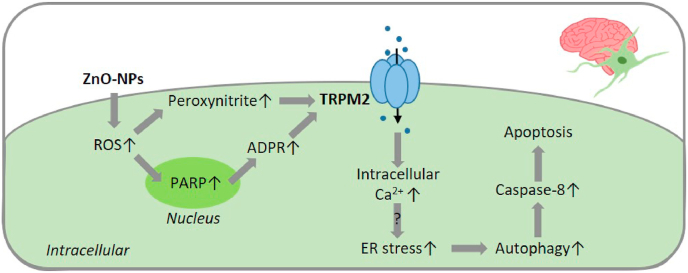
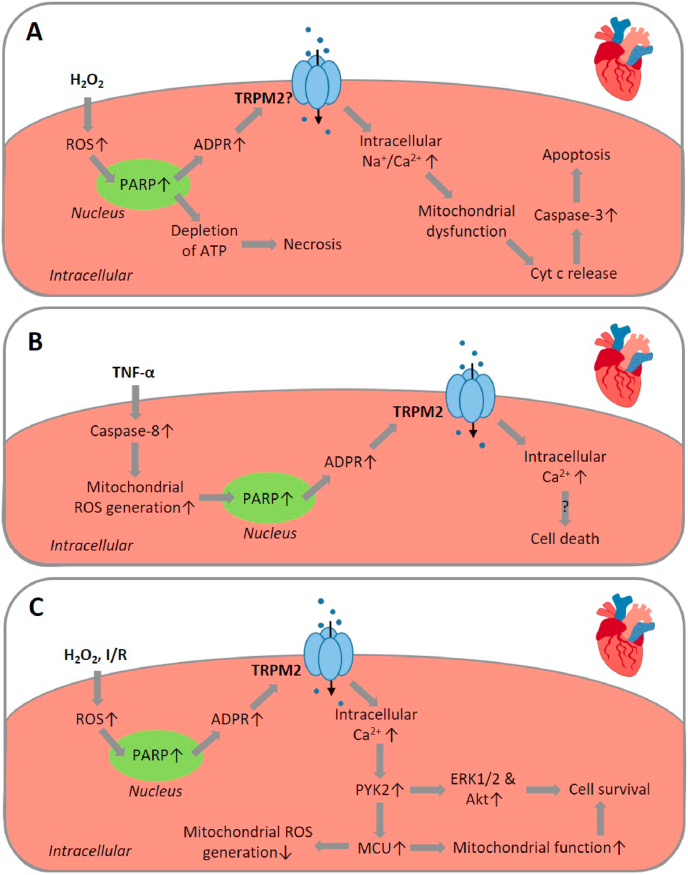
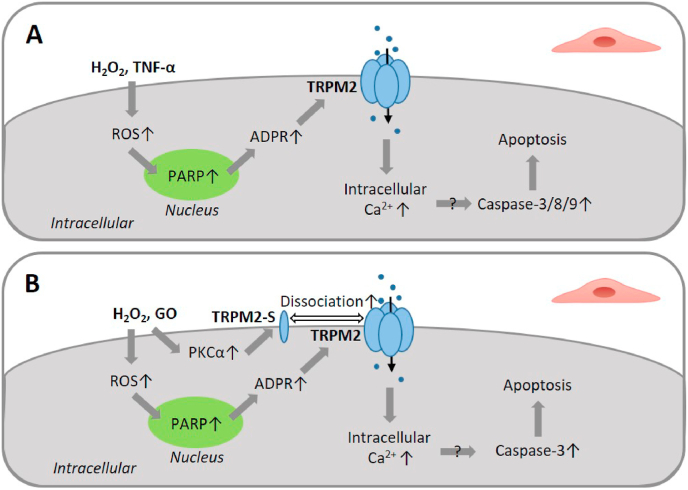
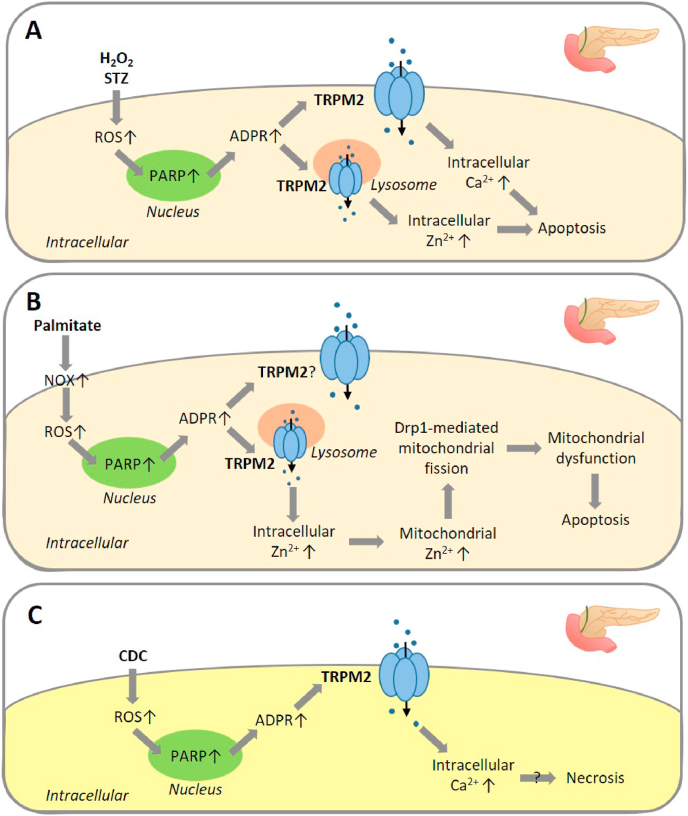
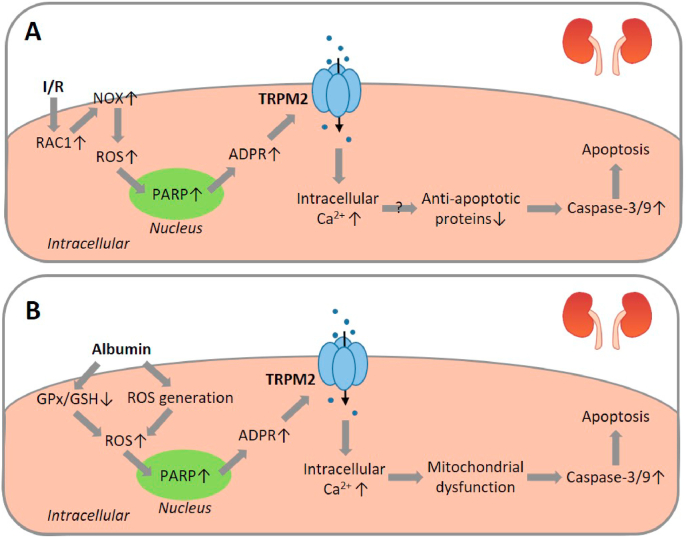
Oxidative stress resulting from the accumulation of high levels of reactive oxygen species is a salient feature of, and a well-recognised pathological factor for, diverse pathologies. One common mechanism for oxidative stress damage is via the disruption of intracellular ion homeostasis to induce cell death. TRPM2 is a non-selective Ca-permeable cation channel with a wide distribution throughout the body and is highly sensitive to activation by oxidative stress. Recent studies have collected abundant evidence to show its important role in mediating cell death induced by miscellaneous oxidative stress-inducing pathological factors, both endogenous and exogenous, including ischemia/reperfusion and the neurotoxicants amyloid-β peptides and MPTP/MPP that cause neuronal demise in the brain, myocardial ischemia/reperfusion, proinflammatory mediators that disrupt endothelial function, diabetogenic agent streptozotocin and diabetes risk factor free fatty acids that induce loss of pancreatic β-cells, bile acids that damage pancreatic acinar cells, renal ischemia/reperfusion and albuminuria that are detrimental to kidney cells, acetaminophen that triggers hepatocyte death, and nanoparticles that injure pericytes. Studies have also shed light on the signalling mechanisms by which these pathological factors activate the TRPM2 channel to alter intracellular ion homeostasis leading to aberrant initiation of various cell death pathways. TRPM2-mediated cell death thus emerges as an important mechanism in the pathogenesis of conditions including ischemic stroke, neurodegenerative diseases, cardiovascular diseases, diabetes, pancreatitis, chronic kidney disease, liver damage and neurovascular injury. These findings raise the exciting perspective of targeting the TRPM2 channel as a novel therapeutic strategy to treat such oxidative stress-associated diseases.
氧化应激是由高水平活性氧物质积累引起的,是多种病理的显著特征和公认的病理因素。氧化应激损伤的一个常见机制是通过破坏细胞内离子稳态来诱导细胞死亡。TRPM2 是一种非选择性钙通透性阳离子通道,在体内广泛分布,对氧化应激的激活非常敏感。最近的研究收集了丰富的证据表明,它在外源和内源多种氧化应激诱导的病理因素(包括缺血再灌注和导致脑神经元死亡的神经毒性物质淀粉样β肽和 MPTP/MPP、心肌缺血再灌注、破坏内皮功能的促炎介质、导致胰岛β细胞丧失的致糖尿病剂链脲佐菌素和糖尿病风险因素游离脂肪酸、损伤胰腺腺泡细胞的胆汁酸、对肾脏细胞有害的肾缺血再灌注和蛋白尿、引发肝细胞死亡的对乙酰氨基酚以及损伤周细胞的纳米颗粒)诱导的细胞死亡中发挥重要作用。这些研究还揭示了这些病理因素激活 TRPM2 通道改变细胞内离子稳态从而导致各种细胞死亡途径异常启动的信号机制。因此,TRPM2 介导的细胞死亡成为包括缺血性中风、神经退行性疾病、心血管疾病、糖尿病、胰腺炎、慢性肾病、肝损伤和神经血管损伤在内的多种疾病发病机制中的一个重要机制。这些发现提出了一个令人兴奋的观点,即靶向 TRPM2 通道作为一种治疗氧化应激相关疾病的新的治疗策略。