Telford Marco, Hughes David A, Juan David, Stoneking Mark, Navarro Arcadi, Santpere Gabriel
Institute of Evolutionary Biology (CSIC-Universitat Pompeu Fabra), Department of Experimental and Health Sciences (DCEXS), Barcelona Biomedical Research Park, 08003 Barcelona, Spain.
Bristol Population Health Science Institute, University of Bristol, Bristol BS8 2BN, UK.
Microorganisms. 2020 Oct 29;8(11):1686. doi: 10.3390/microorganisms8111686.
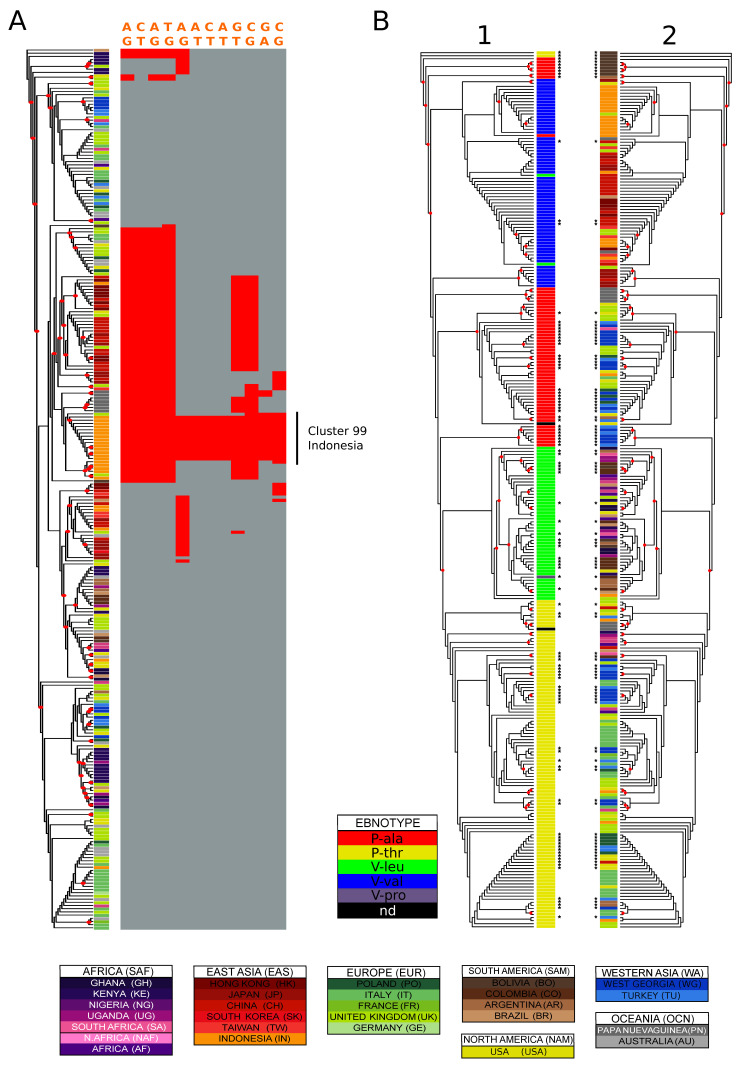
The Epstein-Barr Virus (EBV) infects the vast majority of human individuals worldwide (~90%) and is associated with several diseases, including different types of cancer and multiple sclerosis, which show wide variation in incidence among global geographical regions. Genetic variants in EBV genomic sequences have been used to determine the geographical structure of EBV isolates, but our understanding of EBV diversity remains highly incomplete. We generated sequences for 13 pivotal EBV genes derived from 103 healthy individuals, expanding current EBV diversity datasets with respect to both geographic coverage and number of isolates per region. These newly generated sequences were integrated with the more than 250 published EBV genomes, generating the most geographically comprehensive data set of EBV strains to date. We report remarkable variation in single-gene phylogenies that, when analysed together, show robust signals of population structure. Our results not only confirm known major global patterns of geographic variation, such as the clear separation of Asian isolates from the rest, and the intermixed relationships among African, European and Australian isolates, but yield novel phylogenetic relationships with previously unreported populations. We provide a better understanding of EBV's population structure in South America, Africa and, by the inclusion of Turkey and Georgia, we also gain insight into EBV diversity in Western Asia, a crossroads connecting Europe, Africa and Asia. In summary, our results provide a detailed world-wide characterisation of EBV genetic clusters, their enrichment in specific geographic regions, novel inter-population relationships, and a catalogue of geographically informative EBV genetic variants.
爱泼斯坦-巴尔病毒(EBV)感染了全球绝大多数人(约90%),并与多种疾病相关,包括不同类型的癌症和多发性硬化症,这些疾病在全球不同地理区域的发病率差异很大。EBV基因组序列中的基因变异已被用于确定EBV分离株的地理结构,但我们对EBV多样性的了解仍然非常不完整。我们从103名健康个体中生成了13个关键EBV基因的序列,在地理覆盖范围和每个区域的分离株数量方面扩展了当前的EBV多样性数据集。这些新生成的序列与250多个已发表的EBV基因组整合在一起,生成了迄今为止地理上最全面的EBV菌株数据集。我们报告了单基因系统发育中的显著变异,综合分析时显示出强大的种群结构信号。我们的结果不仅证实了已知的主要全球地理变异模式,如亚洲分离株与其他地区的明显分离,以及非洲、欧洲和澳大利亚分离株之间的混合关系,还产生了与以前未报道群体的新系统发育关系。我们更好地了解了南美洲和非洲的EBV种群结构,通过纳入土耳其和格鲁吉亚,我们还深入了解了西亚的EBV多样性,西亚是连接欧洲、非洲和亚洲的十字路口。总之,我们的结果提供了EBV遗传簇的详细全球特征、它们在特定地理区域的富集情况、新的种群间关系,以及一份具有地理信息的EBV基因变异目录。