Graduate Program in Computational and Data Sciences, Schmid College of Science and Technology, Chapman University, Orange, CA 92866, USA.
Department of Biomedical and Pharmaceutical Sciences, Chapman University School of Pharmacy, Irvine, CA 92618, USA.
Int J Mol Sci. 2020 Nov 4;21(21):8268. doi: 10.3390/ijms21218268.
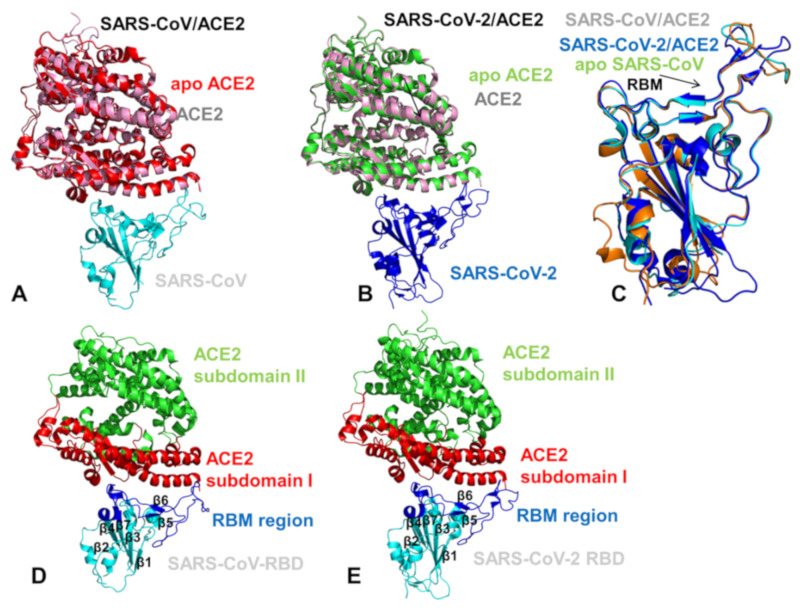
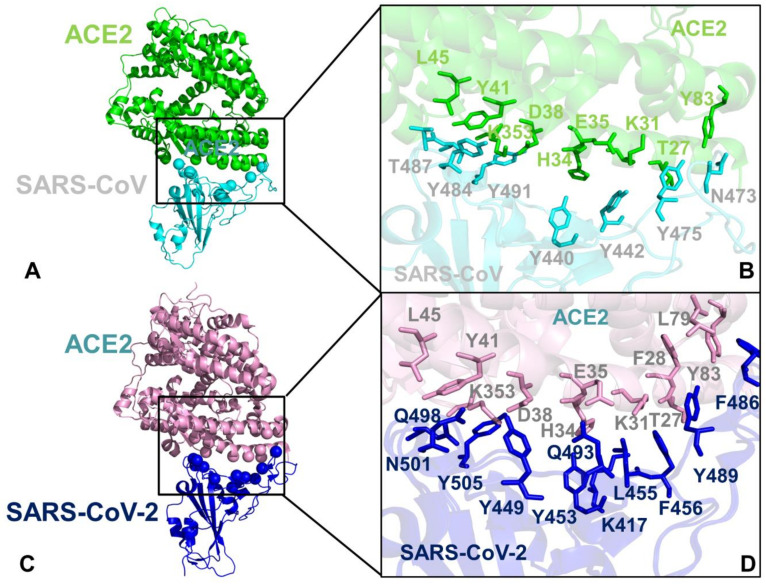
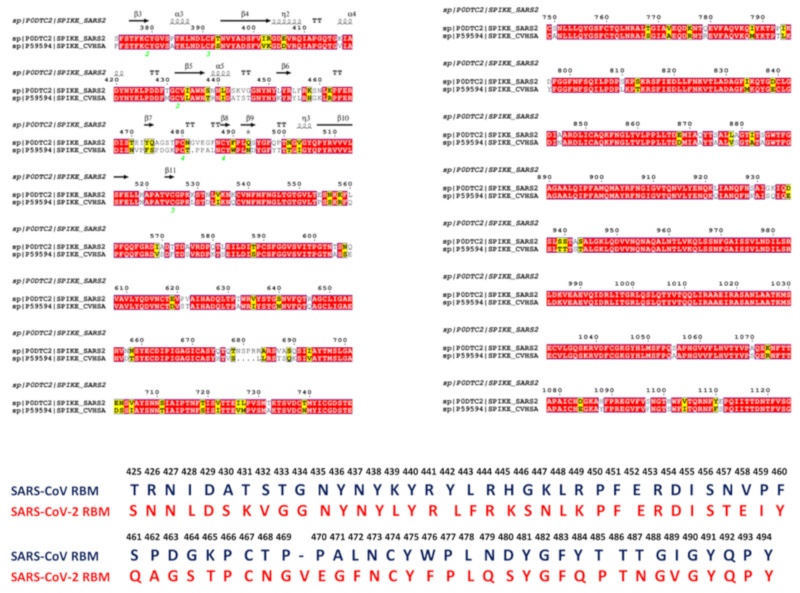
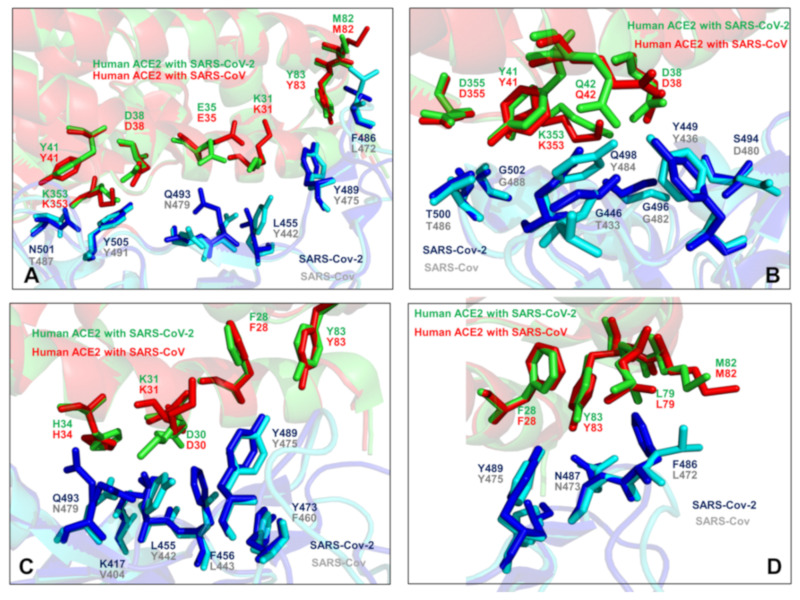
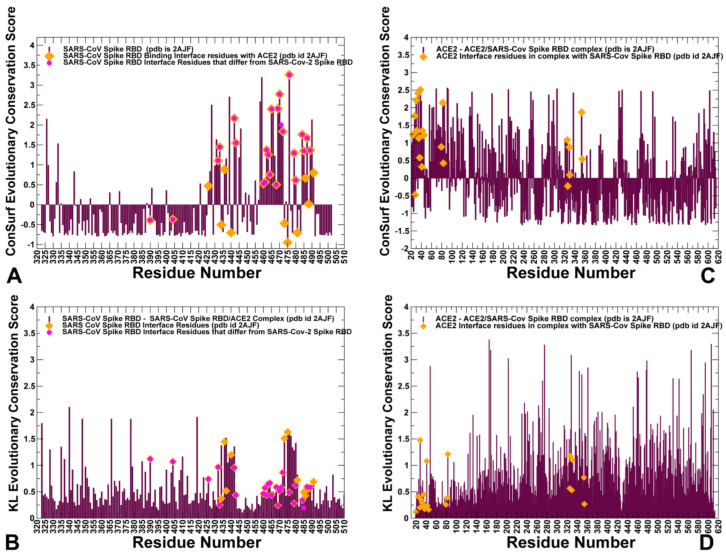
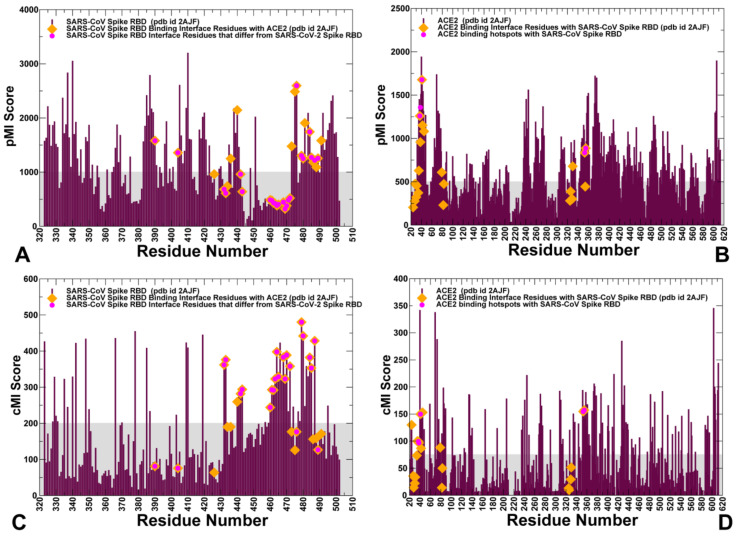
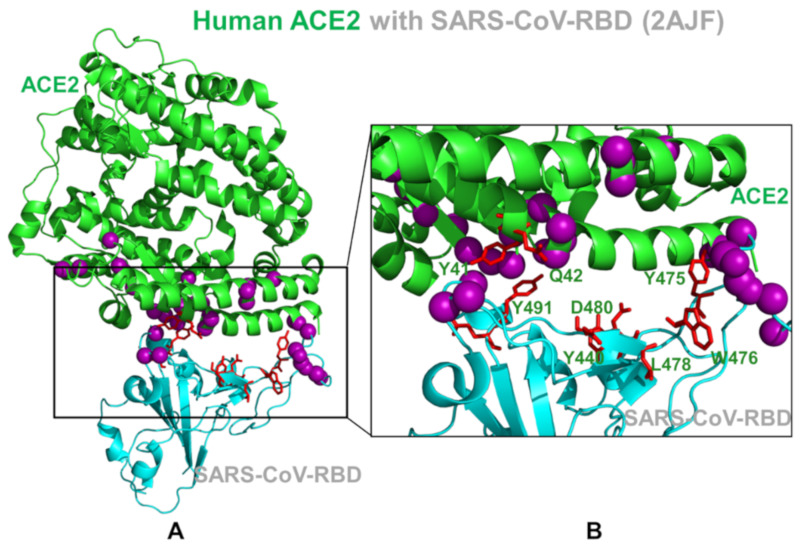
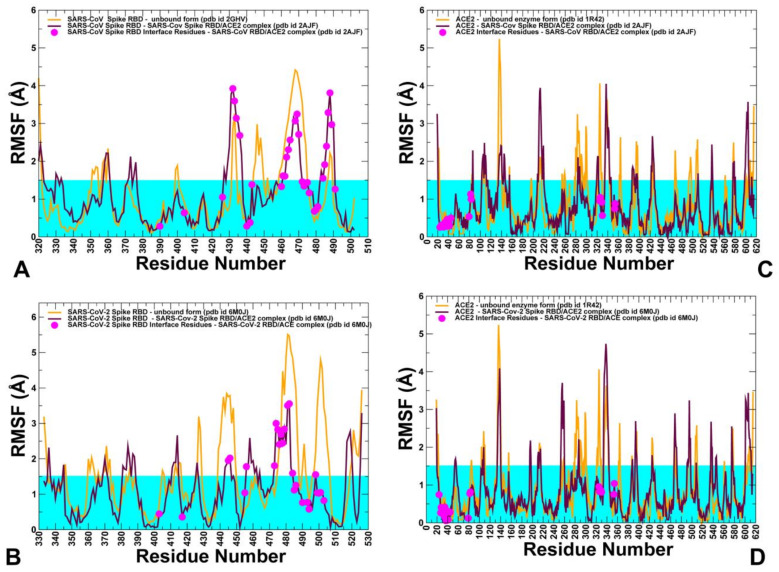
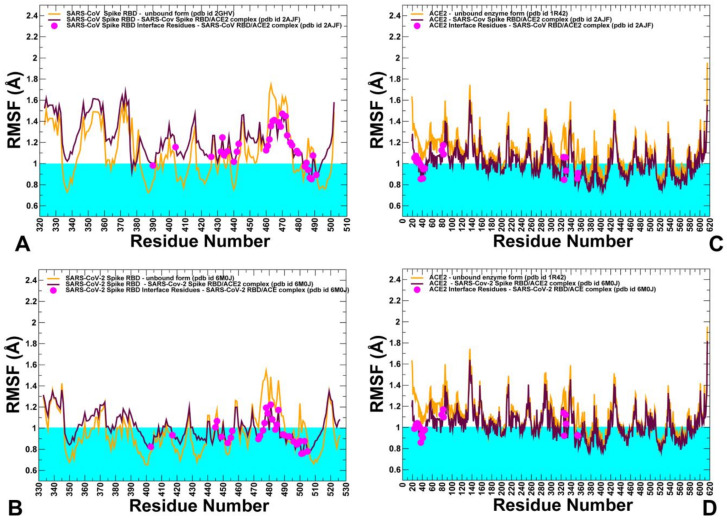
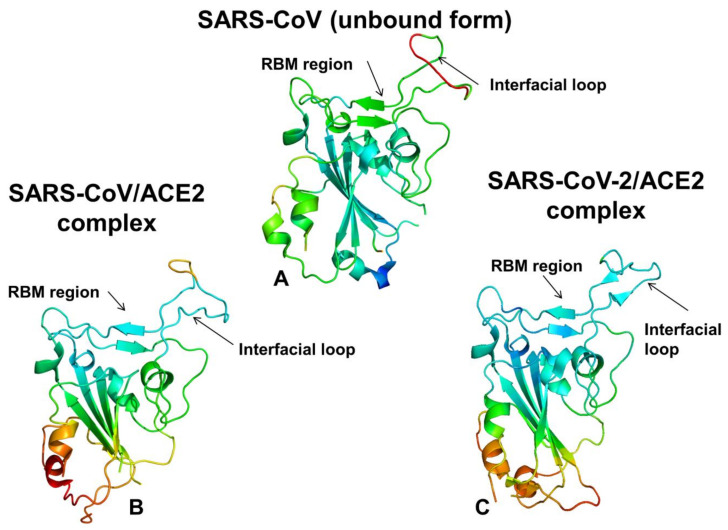
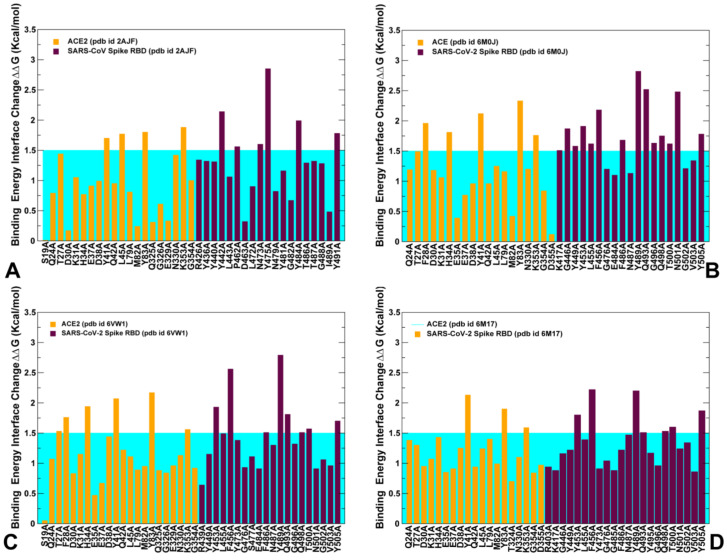
Binding to the host receptor is a critical initial step for the coronavirus SARS-CoV-2 spike protein to enter into target cells and trigger virus transmission. A detailed dynamic and energetic view of the binding mechanisms underlying virus entry is not fully understood and the consensus around the molecular origins behind binding preferences of SARS-CoV-2 for binding with the angiotensin-converting enzyme 2 (ACE2) host receptor is yet to be established. In this work, we performed a comprehensive computational investigation in which sequence analysis and modeling of coevolutionary networks are combined with atomistic molecular simulations and comparative binding free energy analysis of the SARS-CoV and SARS-CoV-2 spike protein receptor binding domains with the ACE2 host receptor. Different from other computational studies, we systematically examine the molecular and energetic determinants of the binding mechanisms between SARS-CoV-2 and ACE2 proteins through the lens of coevolution, conformational dynamics, and allosteric interactions that conspire to drive binding interactions and signal transmission. Conformational dynamics analysis revealed the important differences in mobility of the binding interfaces for the SARS-CoV-2 spike protein that are not confined to several binding hotspots, but instead are broadly distributed across many interface residues. Through coevolutionary network analysis and dynamics-based alanine scanning, we established linkages between the binding energy hotspots and potential regulators and carriers of signal communication in the virus-host receptor complexes. The results of this study detailed a binding mechanism in which the energetics of the SARS-CoV-2 association with ACE2 may be determined by cumulative changes of a number of residues distributed across the entire binding interface. The central findings of this study are consistent with structural and biochemical data and highlight drug discovery challenges of inhibiting large and adaptive protein-protein interfaces responsible for virus entry and infection transmission.
冠状病毒 SARS-CoV-2 刺突蛋白与宿主受体结合是其进入靶细胞并引发病毒传播的关键初始步骤。目前,人们对于病毒进入的结合机制的动态和能量学观点还没有完全理解,对于 SARS-CoV-2 与血管紧张素转化酶 2(ACE2)宿主受体结合偏好的分子起源也没有达成共识。在这项工作中,我们进行了全面的计算研究,将共进化网络的序列分析和建模与原子分子模拟以及 SARS-CoV 和 SARS-CoV-2 刺突蛋白受体结合域与 ACE2 宿主受体的比较结合自由能分析相结合。与其他计算研究不同的是,我们通过共进化、构象动力学和变构相互作用的视角系统地研究了 SARS-CoV-2 和 ACE2 蛋白之间的结合机制的分子和能量决定因素,这些因素共同驱动结合相互作用和信号转导。构象动力学分析揭示了 SARS-CoV-2 刺突蛋白结合界面的重要差异,这些差异不仅局限于几个结合热点,而是广泛分布在许多界面残基上。通过共进化网络分析和基于动力学的丙氨酸扫描,我们建立了病毒-宿主受体复合物中结合能热点与潜在信号转导调节剂和载体之间的联系。这项研究的结果详细描述了一种结合机制,即 SARS-CoV-2 与 ACE2 结合的能量可能由分布在整个结合界面上的许多残基的累积变化决定。本研究的主要结论与结构和生化数据一致,并强调了抑制负责病毒进入和感染传播的大型和适应性蛋白-蛋白界面的药物发现挑战。