Hess Matthias, Paul Shyam S, Puniya Anil K, van der Giezen Mark, Shaw Claire, Edwards Joan E, Fliegerová Kateřina
Systems Microbiology & Natural Product Discovery Laboratory, Department of Animal Science, University of California, Davis, Davis, CA, United States.
Gut Microbiome Lab, ICAR-Directorate of Poultry Research, Indian Council of Agricultural Research, Hyderabad, India.
Front Microbiol. 2020 Oct 21;11:584893. doi: 10.3389/fmicb.2020.584893. eCollection 2020.
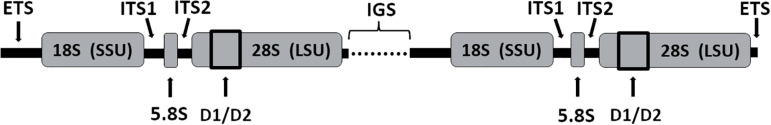
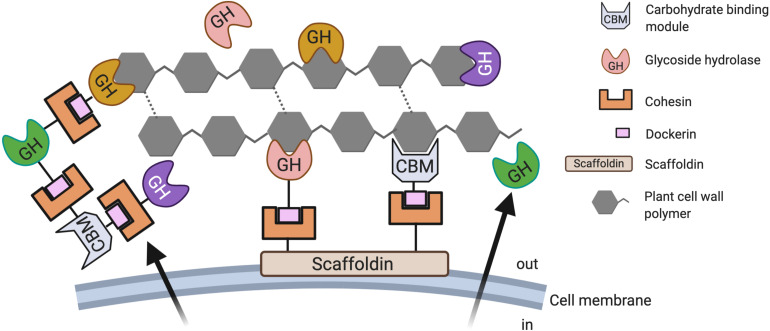
Anaerobic fungi (AF) play an essential role in feed conversion due to their potent fiber degrading enzymes and invasive growth. Much has been learned about this unusual fungal phylum since the paradigm shifting work of Colin Orpin in the 1970s, when he characterized the first AF. Molecular approaches targeting specific phylogenetic marker genes have facilitated taxonomic classification of AF, which had been previously been complicated by the complex life cycles and associated morphologies. Although we now have a much better understanding of their diversity, it is believed that there are still numerous genera of AF that remain to be described in gut ecosystems. Recent marker-gene based studies have shown that fungal diversity in the herbivore gut is much like the bacterial population, driven by host phylogeny, host genetics and diet. Since AF are major contributors to the degradation of plant material ingested by the host animal, it is understandable that there has been great interest in exploring the enzymatic repertoire of these microorganisms in order to establish a better understanding of how AF, and their enzymes, can be used to improve host health and performance, while simultaneously reducing the ecological footprint of the livestock industry. A detailed understanding of AF and their interaction with other gut microbes as well as the host animal is essential, especially when production of affordable high-quality protein and other animal-based products needs to meet the demands of an increasing human population. Such a mechanistic understanding, leading to more sustainable livestock practices, will be possible with recently developed -omics technologies that have already provided first insights into the different contributions of the fungal and bacterial population in the rumen during plant cell wall hydrolysis.
厌氧真菌(AF)由于其强大的纤维降解酶和侵入性生长,在饲料转化中起着至关重要的作用。自20世纪70年代科林·奥尔平(Colin Orpin)的开创性工作以来,我们对这个不同寻常的真菌门有了很多了解,当时他对首个厌氧真菌进行了特征描述。针对特定系统发育标记基因的分子方法促进了厌氧真菌的分类,此前其复杂的生命周期和相关形态使分类工作变得复杂。尽管我们现在对它们的多样性有了更好的理解,但据信在肠道生态系统中仍有许多厌氧真菌属有待描述。最近基于标记基因的研究表明,食草动物肠道中的真菌多样性与细菌群体非常相似,受宿主系统发育、宿主遗传学和饮食的驱动。由于厌氧真菌是宿主动物摄入的植物材料降解的主要贡献者,因此人们对探索这些微生物的酶库产生了浓厚兴趣,以便更好地理解厌氧真菌及其酶如何用于改善宿主健康和性能,同时减少畜牧业的生态足迹。详细了解厌氧真菌及其与其他肠道微生物以及宿主动物的相互作用至关重要,特别是当生产负担得起的高质量蛋白质和其他动物产品需要满足不断增长的人口需求时。借助最近开发的组学技术,这种有助于实现更可持续畜牧实践的机制性理解将成为可能,这些技术已经为瘤胃中真菌和细菌群体在植物细胞壁水解过程中的不同贡献提供了初步见解。