Department of Molecular Biology and Genetics, Center for Quantitative Genetics and Genomics, Aarhus University, Aarhus, Denmark.
Bioinformatics Research Centre, Aarhus University, Aarhus, Denmark.
ISME J. 2020 Aug;14(8):2019-2033. doi: 10.1038/s41396-020-0663-x. Epub 2020 May 4.
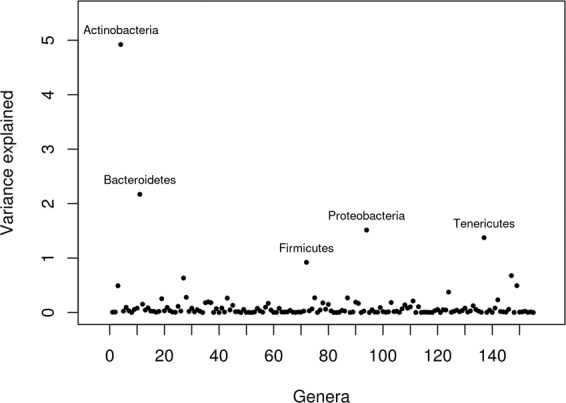
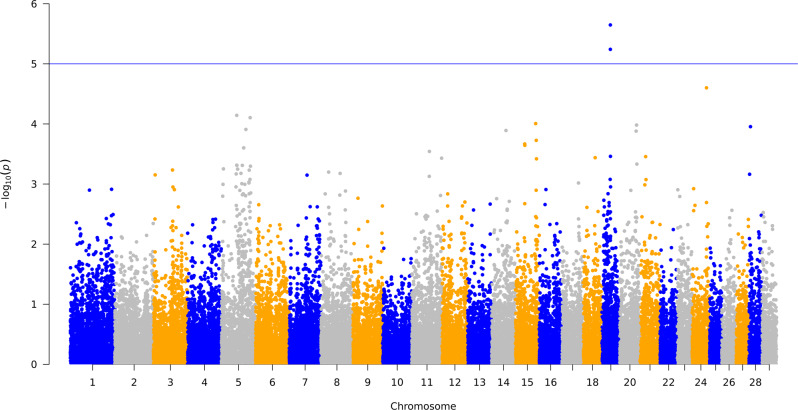
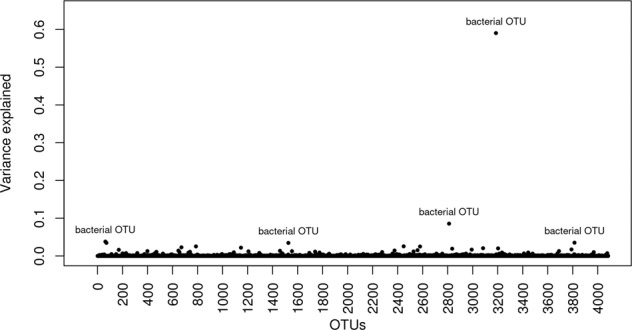
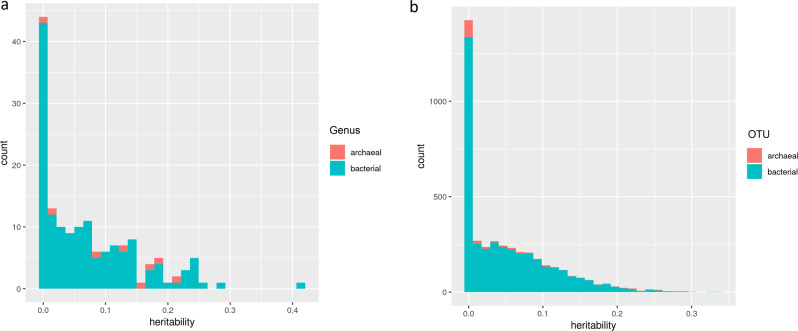
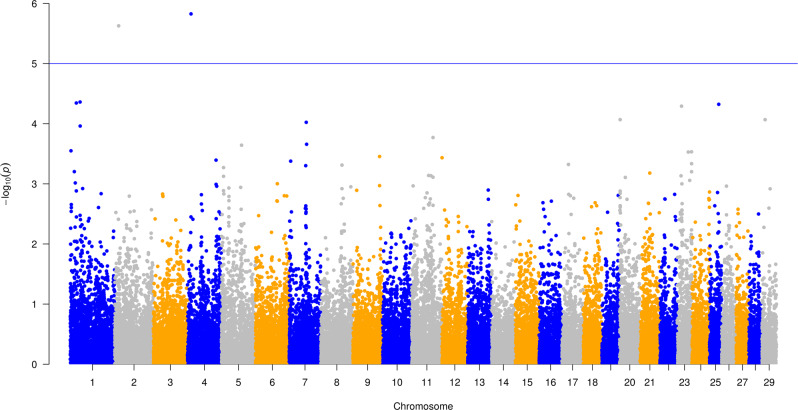
Reducing methane emissions from livestock production is of great importance for the sustainable management of the Earth's environment. Rumen microbiota play an important role in producing biogenic methane. However, knowledge of how host genetics influences variation in ruminal microbiota and their joint effects on methane emission is limited. We analyzed data from 750 dairy cows, using a Bayesian model to simultaneously assess the impact of host genetics and microbiota on host methane emission. We estimated that host genetics and microbiota explained 24% and 7%, respectively, of variation in host methane levels. In this Bayesian model, one bacterial genus explained up to 1.6% of the total microbiota variance. Further analysis was performed by a mixed linear model to estimate variance explained by host genomics in abundances of microbial genera and operational taxonomic units (OTU). Highest estimates were observed for a bacterial OTU with 33%, for an archaeal OTU with 26%, and for a microbial genus with 41% heritability. However, after multiple testing correction for the number of genera and OTUs modeled, none of the effects remained significant. We also used a mixed linear model to test effects of individual host genetic markers on microbial genera and OTUs. In this analysis, genetic markers inside host genes ABS4 and DNAJC10 were found associated with microbiota composition. We show that a Bayesian model can be utilized to model complex structure and relationship between microbiota simultaneously and their interaction with host genetics on methane emission. The host genome explains a significant fraction of between-individual variation in microbial abundance. Individual microbial taxonomic groups each only explain a small amount of variation in methane emissions. The identification of genes and genetic markers suggests that it is possible to design strategies for breeding cows with desired microbiota composition associated with phenotypes.
减少畜牧业甲烷排放对地球环境的可持续管理至关重要。瘤胃微生物群在产生生物甲烷方面发挥着重要作用。然而,宿主遗传学如何影响瘤胃微生物群的变异及其对甲烷排放的共同影响的知识有限。我们分析了 750 头奶牛的数据,使用贝叶斯模型同时评估宿主遗传学和微生物群对宿主甲烷排放的影响。我们估计宿主遗传学和微生物群分别解释了宿主甲烷水平变异的 24%和 7%。在这个贝叶斯模型中,一个细菌属解释了多达 1.6%的总微生物群方差。通过混合线性模型进一步分析,以估计宿主基因组学在微生物属和操作分类单元(OTU)丰度上解释的方差。对细菌 OTU 观察到的最高估计值为 33%,对古菌 OTU 为 26%,对微生物属为 41%。然而,在对建模的属和 OTU 数量进行多次测试校正后,没有一个效应仍然显著。我们还使用混合线性模型来测试个体宿主遗传标记对微生物属和 OTU 的影响。在这项分析中,发现宿主基因 ABS4 和 DNAJC10 内的遗传标记与微生物群落组成相关。我们表明,贝叶斯模型可用于同时模拟微生物群落的复杂结构和关系及其与宿主遗传学对甲烷排放的相互作用。宿主基因组解释了个体间微生物丰度变异的很大一部分。每个个体微生物分类群仅解释了甲烷排放的一小部分变异。基因和遗传标记的鉴定表明,有可能设计出与表型相关的具有所需微生物群落组成的奶牛育种策略。