Gao Shan, Chen Tianxiang, Li Lijie, Liu Xin, Liu Yang, Zhao Junjun, Lu Qiliang, Zeng Zhi, Xu Qiuran, Huang Dongsheng, Tu Kangsheng
Key Laboratory of Tumor Molecular Diagnosis and Individualized Medicine of Zhejiang Province, Zhejiang Provincial People's Hospital (People's Hospital of Hangzhou Medical College), Hangzhou, China.
The Second Clinical Medical College, Zhejiang Chinese Medical University, Hangzhou, China.
Front Cell Dev Biol. 2020 Oct 19;8:587389. doi: 10.3389/fcell.2020.587389. eCollection 2020.
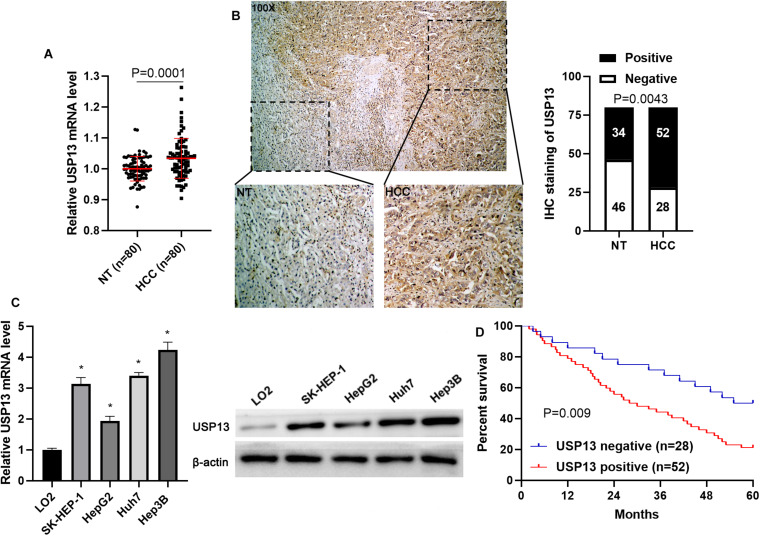
Hepatocellular carcinoma (HCC) is one of the leading causes of cancer death worldwide. The activation of the toll-like receptor 4/myeloid differentiation primary response gene 88/nuclear factor-κB (TLR4/MyD88/NF-κB) pathway contributes to the development and progression of HCC. The ubiquitin-proteasome system regulates TLR4 expression. However, whether ubiquitin specific peptidase 13 (USP13) stabilizes TLR4 and facilitates HCC progression remains unclear. Here, quantitative real-time PCR (qRT-PCR) and immunohistochemistry analysis revealed that USP13 expression in HCC tissues was higher than in non-tumor liver tissues. Moreover, the elevated expression of USP13 was detected in HCC cells (SK-HEP-1, HepG2, Huh7, and Hep3B) compared to LO2 cells. Interestingly, the positive staining of USP13 was closely correlated with tumor size ≥ 5 cm and advanced tumor stage and conferred to significantly lower survival of HCC patients. Next, USP13 knockdown prominently reduced the proliferation, epithelial-mesenchymal transition (EMT), migration, and invasion of Hep3B and Huh7 cells, while USP13 overexpression enhanced these biological behaviors of HepG2 and LO2 cells. The silencing of USP13 significantly restrained the growth and lung metastasis of HCC cells . Mechanistically, the USP13 depletion markedly inhibited the TLR4/MyD88/NF-κB pathway in HCC cells. USP13 interacted with TLR4 and inhibited the ubiquitin-mediated degradation of TLR4. Significantly, TLR4 re-expression remarkably reversed the effects of USP13 knockdown on HCC cells. USP13 expression was markedly upregulated in HCC cells under hypoxia conditions. Notably, USP13 knockdown repressed hypoxia-induced activation of the TLR4/MyD88/NF-κB pathway in HCC cells. In conclusion, our study uncovered that hypoxia-induced USP13 facilitated HCC progression via enhancing TLR4 deubiquitination and subsequently activating the TLR4/MyD88/NF-κB pathway.
肝细胞癌(HCC)是全球癌症死亡的主要原因之一。Toll样受体4/髓样分化初级反应基因88/核因子-κB(TLR4/MyD88/NF-κB)信号通路的激活促进了HCC的发生和发展。泛素-蛋白酶体系统调节TLR4的表达。然而,泛素特异性肽酶13(USP13)是否能稳定TLR4并促进HCC进展仍不清楚。在此,定量实时PCR(qRT-PCR)和免疫组化分析显示,HCC组织中USP13的表达高于非肿瘤肝组织。此外,与LO2细胞相比,在HCC细胞(SK-HEP-1、HepG2、Huh7和Hep3B)中检测到USP13表达升高。有趣的是,USP13的阳性染色与肿瘤大小≥5 cm和肿瘤晚期密切相关,且与HCC患者的生存率显著降低有关。接下来,敲低USP13显著降低了Hep3B和Huh7细胞的增殖、上皮-间质转化(EMT)、迁移和侵袭,而USP13过表达增强了HepG2和LO2细胞的这些生物学行为。沉默USP13显著抑制了HCC细胞的生长和肺转移。机制上,USP13的缺失显著抑制了HCC细胞中TLR4/MyD88/NF-κB信号通路。USP13与TLR4相互作用并抑制泛素介导的TLR4降解。值得注意的是,TLR4的重新表达显著逆转了敲低USP13对HCC细胞的影响。在缺氧条件下,HCC细胞中USP13的表达明显上调。值得注意的是,敲低USP13可抑制缺氧诱导的HCC细胞中TLR4/MyD88/NF-κB信号通路的激活。总之,我们的研究发现,缺氧诱导的USP13通过增强TLR4去泛素化并随后激活TLR4/MyD88/NF-κB信号通路促进了HCC的进展。