Laboratory of Neuropharmacology, College of Pharmacy and Gachon Institute of Pharmaceutical Sciences, Gachon University, Incheon 21936, Korea.
Int J Mol Sci. 2020 Nov 14;21(22):8595. doi: 10.3390/ijms21228595.
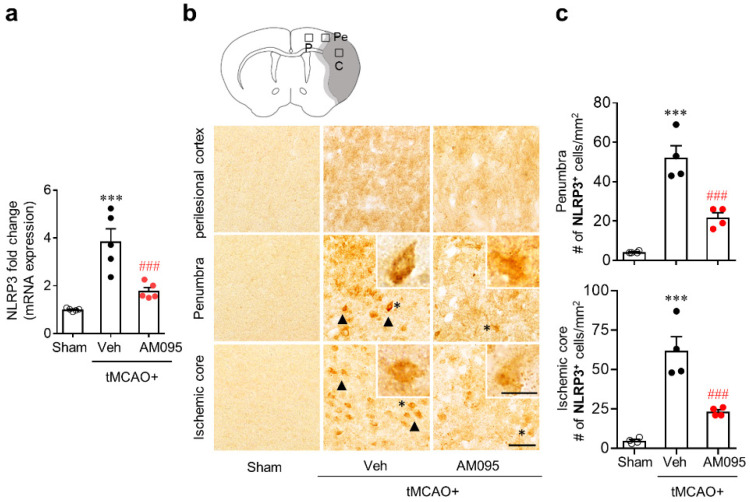
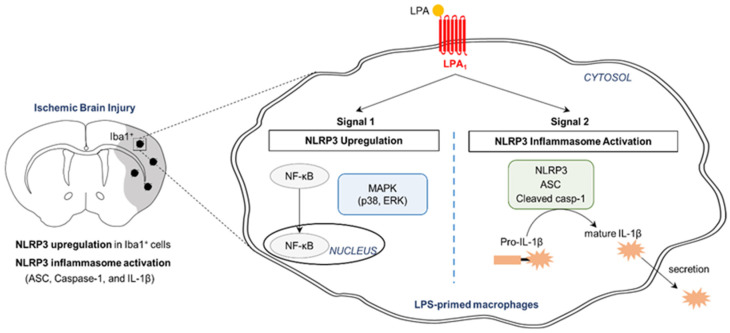
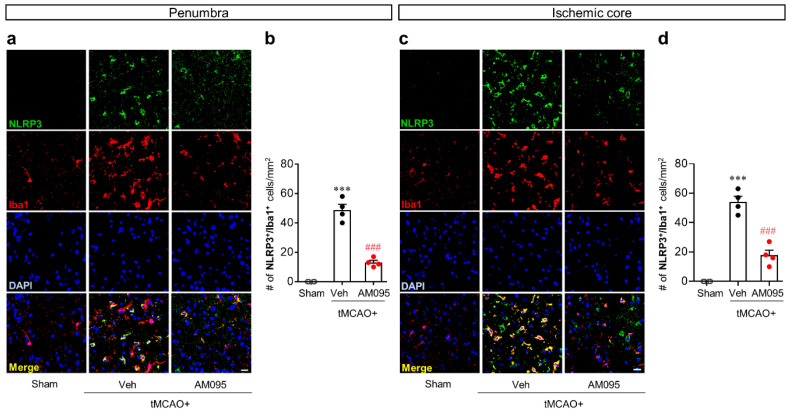
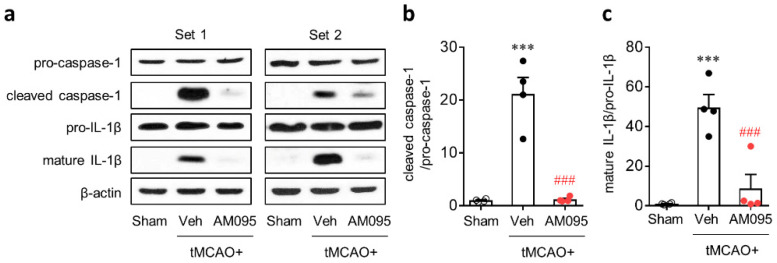
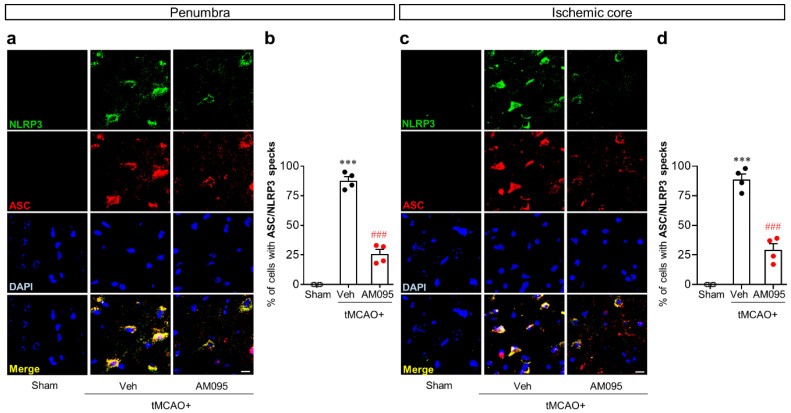
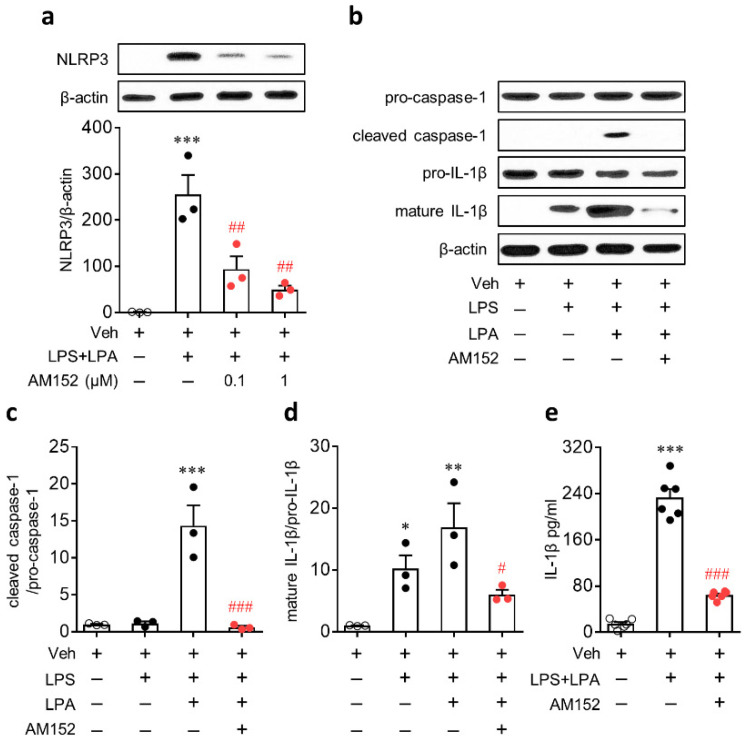
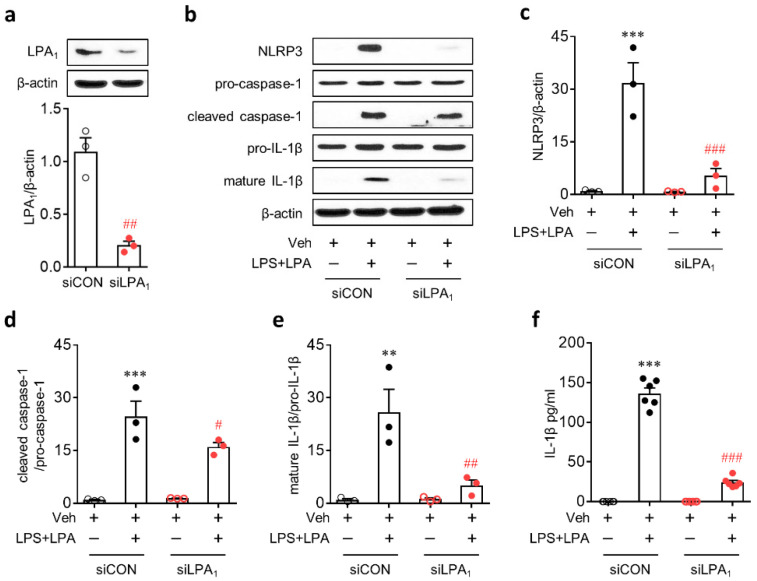
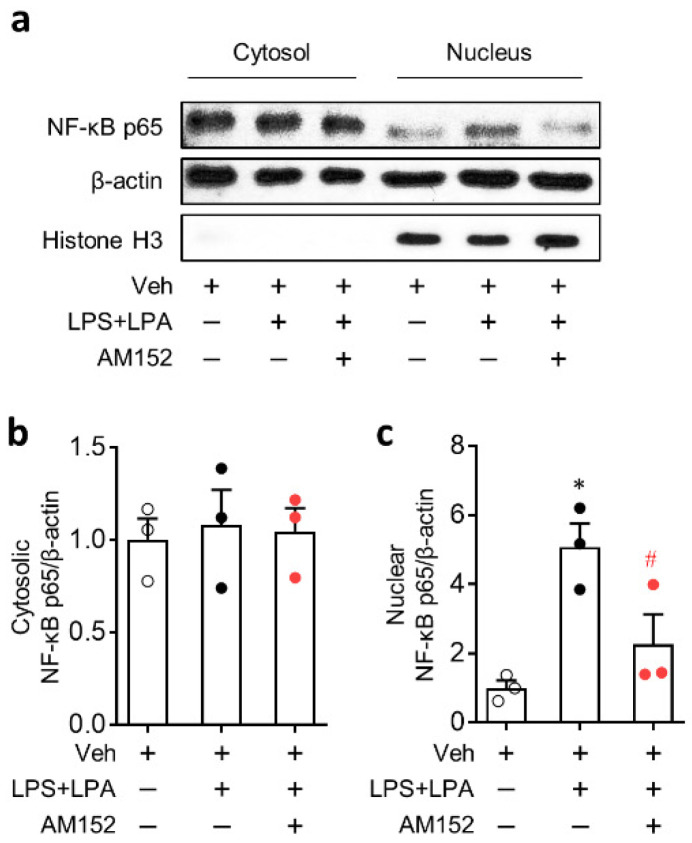
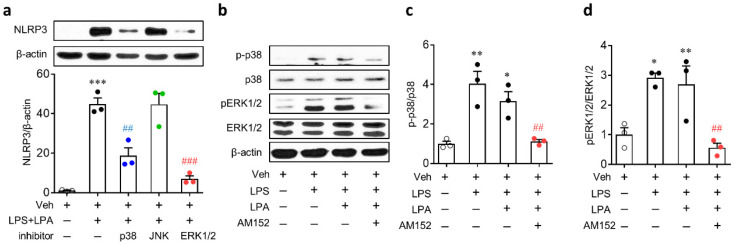
Lysophosphatidic acid receptor 1 (LPA) contributes to brain injury following transient focal cerebral ischemia. However, the mechanism remains unclear. Here, we investigated whether nucleotide-binding oligomerization domain-like receptor family pyrin domain containing 3 (NLRP3) inflammasome activation might be an underlying mechanism involved in the pathogenesis of brain injury associated with LPA following ischemic challenge with transient middle cerebral artery occlusion (tMCAO). Suppressing LPA activity by its antagonist attenuated NLRP3 upregulation in the penumbra and ischemic core regions, particularly in ionized calcium-binding adapter molecule 1 (Iba1)-expressing cells like macrophages of mouse after tMCAO challenge. It also suppressed NLRP3 inflammasome activation, such as caspase-1 activation, interleukin 1β (IL-1β) maturation, and apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) speck formation, in a post-ischemic brain. The role of LPA in NLRP3 inflammasome activation was confirmed in vitro using lipopolysaccharide-primed bone marrow-derived macrophages, followed by LPA exposure. Suppressing LPA activity by either pharmacological antagonism or genetic knockdown attenuated NLRP3 upregulation, caspase-1 activation, IL-1β maturation, and IL-1β secretion in these cells. Furthermore, nuclear factor-κB (NF-κB), extracellular signal-regulated kinase 1/2 (ERK1/2), and p38 were found to be LPA-dependent effector pathways in these cells. Collectively, results of the current study first demonstrate that LPA could contribute to ischemic brain injury by activating NLRP3 inflammasome with underlying effector mechanisms.
溶血磷脂酸受体 1(LPA)在短暂性局灶性脑缺血后导致脑损伤。然而,其机制尚不清楚。在这里,我们研究了核苷酸结合寡聚化结构域样受体家族富含pyrin 结构域 3(NLRP3)炎症小体的激活是否是与缺血性挑战后 LPA 相关的脑损伤发病机制中的潜在机制,短暂性大脑中动脉闭塞(tMCAO)。其拮抗剂抑制 LPA 活性可减轻缺血半影区和缺血核心区 NLRP3 的上调,尤其是在 tMCAO 后,离子钙结合接头分子 1(Iba1)表达细胞如巨噬细胞中。它还抑制 NLRP3 炎症小体的激活,如半胱氨酸蛋白酶-1 的激活、白细胞介素 1β(IL-1β)成熟和凋亡相关斑点样蛋白包含半胱氨酸蛋白酶募集结构域(ASC)斑点形成,在缺血后的大脑中。在体外使用脂多糖预处理的骨髓来源的巨噬细胞,然后暴露于 LPA 后,证实了 LPA 在 NLRP3 炎症小体激活中的作用。通过药理学拮抗或基因敲低抑制 LPA 活性可减弱这些细胞中 NLRP3 的上调、半胱氨酸蛋白酶-1 的激活、IL-1β 的成熟和 IL-1β 的分泌。此外,还发现核因子-κB(NF-κB)、细胞外信号调节激酶 1/2(ERK1/2)和 p38 是这些细胞中 LPA 依赖的效应途径。总之,本研究首次表明,LPA 通过激活 NLRP3 炎症小体并通过潜在的效应机制导致缺血性脑损伤。