School of Computer Science and Engineering, Central South University, Changsha, China.
Division of Biomedical Engineering and Department of Mechanical Engineering, University of Saskatchewan, Saskatoon, SKS7N5A9, Canada.
BMC Bioinformatics. 2020 Nov 18;21(Suppl 6):403. doi: 10.1186/s12859-020-03734-9.
DNA methylation in the human genome is acknowledged to be widely associated with biological processes and complex diseases. The Illumina Infinium methylation arrays have been approved as one of the most efficient and universal technologies to investigate the whole genome changes of methylation patterns. As methylation arrays may still be the dominant method for detecting methylation in the anticipated future, it is crucial to develop a reliable workflow to analysis methylation array data.
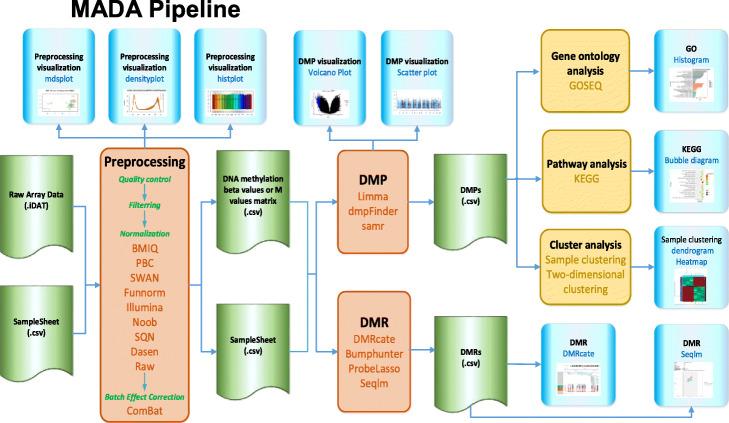
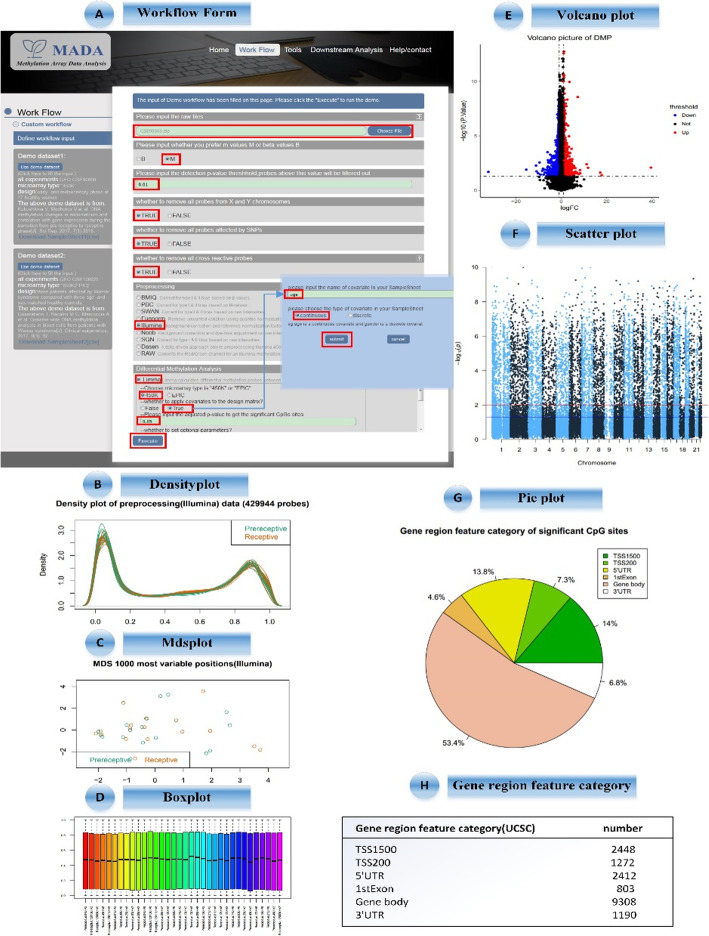
In this study, we develop a web service MADA for the whole process of methylation arrays data analysis, which includes the steps of a comprehensive differential methylation analysis pipeline: pre-processing (data loading, quality control, data filtering, and normalization), batch effect correction, differential methylation analysis, and downstream analysis. In addition, we provide the visualization of pre-processing, differentially methylated probes or regions, gene ontology, pathway and cluster analysis results. Moreover, a customization function for users to define their own workflow is also provided in MADA.
With the analysis of two case studies, we have shown that MADA can complete the whole procedure of methylation array data analysis. MADA provides a graphical user interface and enables users with no computational skills and limited bioinformatics background to carry on complicated methylation array data analysis. The web server is available at: http://120.24.94.89:8080/MADA.
人类基因组中的 DNA 甲基化被广泛认为与生物过程和复杂疾病有关。Illumina Infinium 甲基化芯片已被批准为研究甲基化模式全基因组变化的最有效和通用技术之一。由于在预期的未来,甲基化芯片可能仍然是检测甲基化的主要方法,因此开发一种可靠的工作流程来分析甲基化芯片数据至关重要。
在这项研究中,我们开发了一个名为 MADA 的网络服务,用于甲基化芯片数据分析的全过程,其中包括综合差异甲基化分析管道的步骤:预处理(数据加载、质量控制、数据过滤和归一化)、批次效应校正、差异甲基化分析和下游分析。此外,我们还提供了预处理、差异甲基化探针或区域、基因本体、通路和聚类分析结果的可视化。此外,MADA 还为用户提供了自定义工作流程的功能。
通过对两个案例研究的分析,我们表明 MADA 可以完成甲基化芯片数据分析的整个过程。MADA 提供了一个图形用户界面,使用户即使没有计算技能和有限的生物信息学背景,也能够进行复杂的甲基化芯片数据分析。该网络服务器可在:http://120.24.94.89:8080/MADA 访问。