Institute of Theoretical Chemistry, Faculty of Chemistry, University of Vienna, Währinger Strasse 17, 1090 Vienna, Austria.
Vienna Research Platform on Accelerating Photoreaction Discovery, University of Vienna, Währinger Strasse 17, 1090 Vienna, Austria.
Chem Rev. 2021 Aug 25;121(16):9873-9926. doi: 10.1021/acs.chemrev.0c00749. Epub 2020 Nov 19.
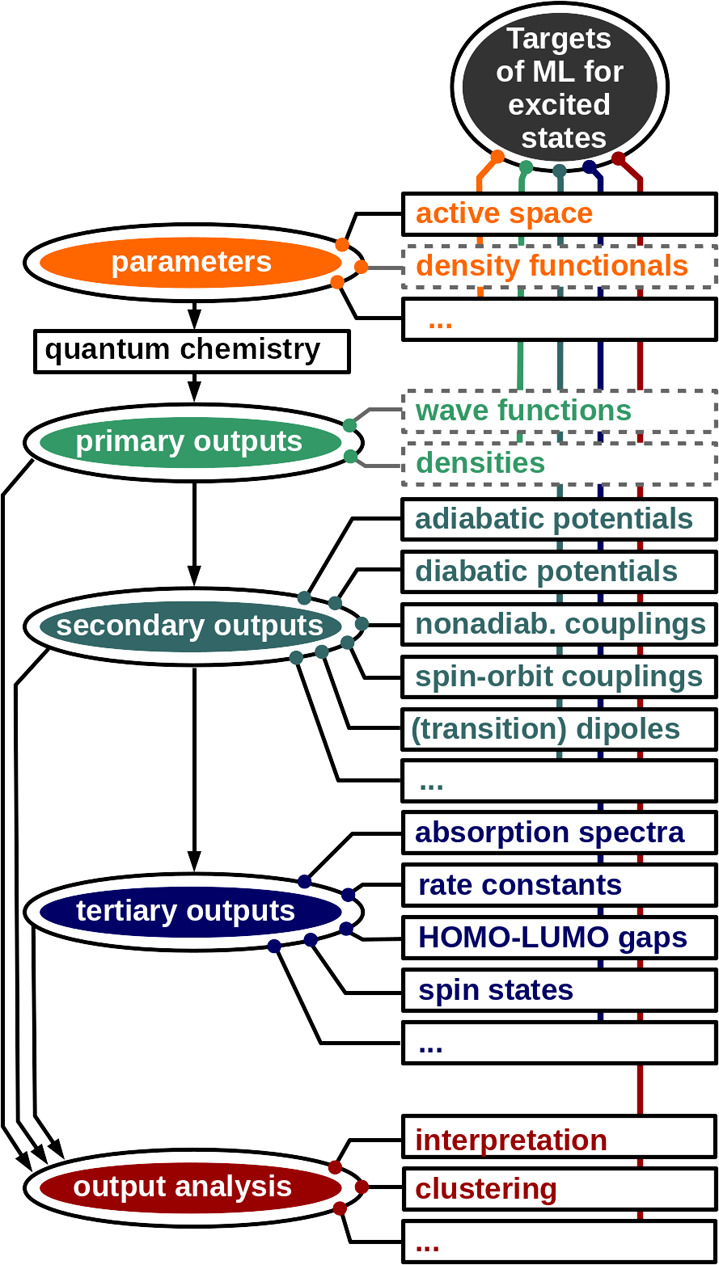
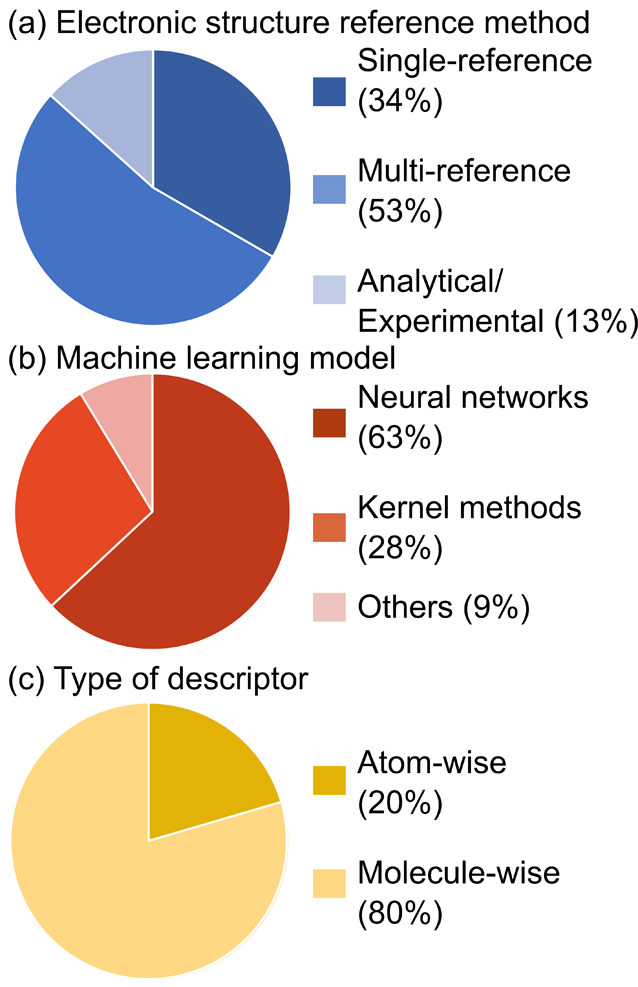
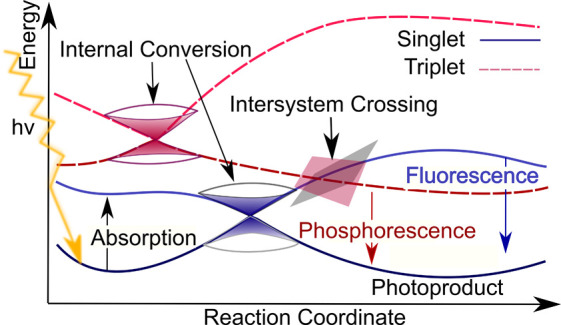
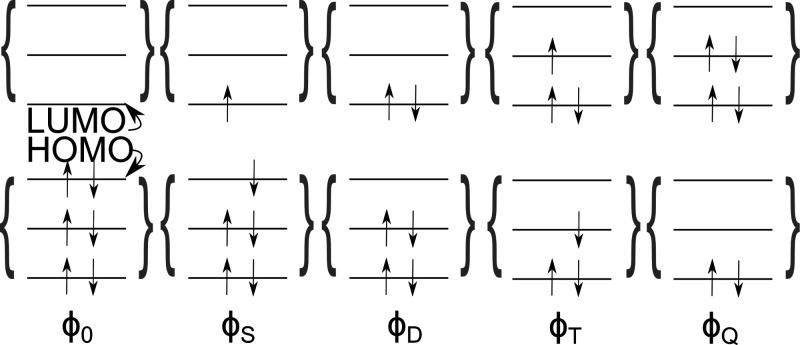
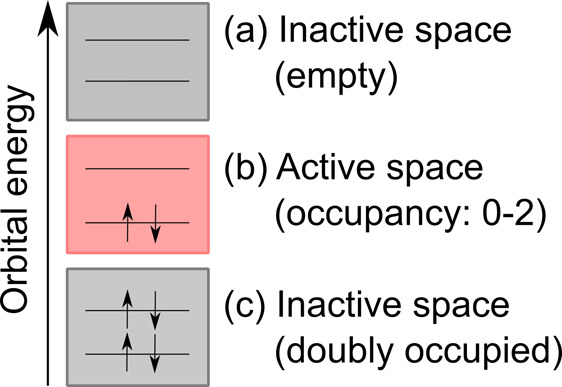
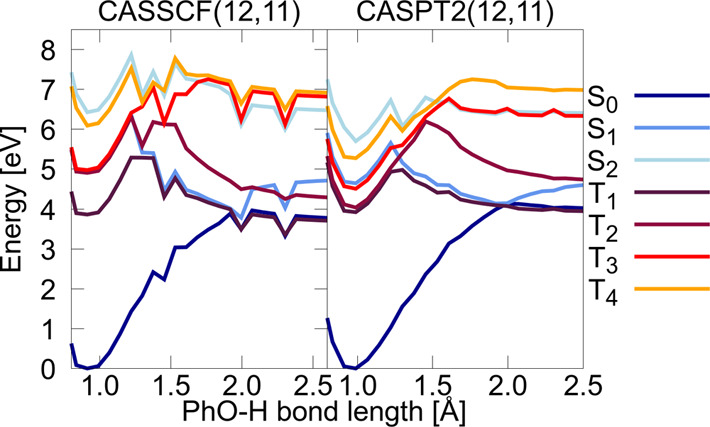
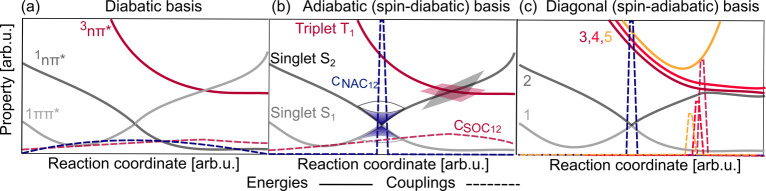
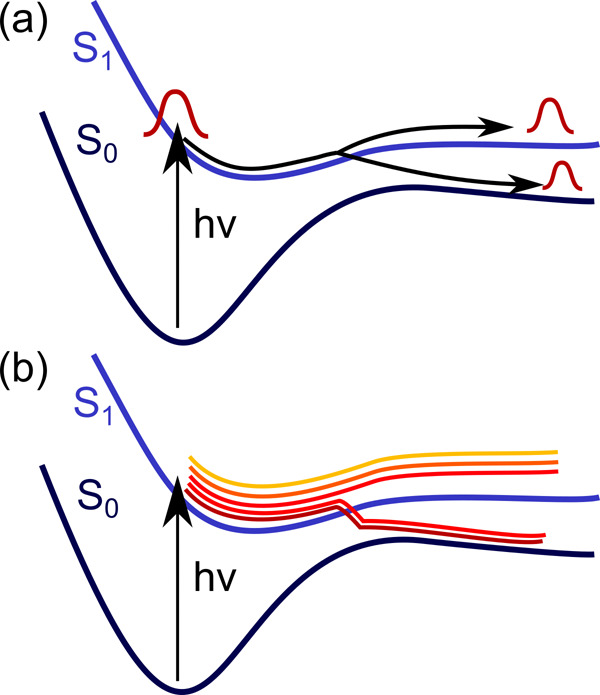
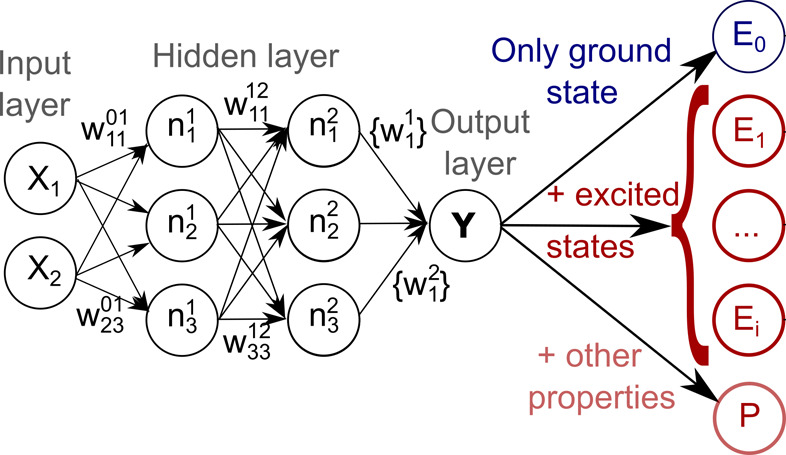
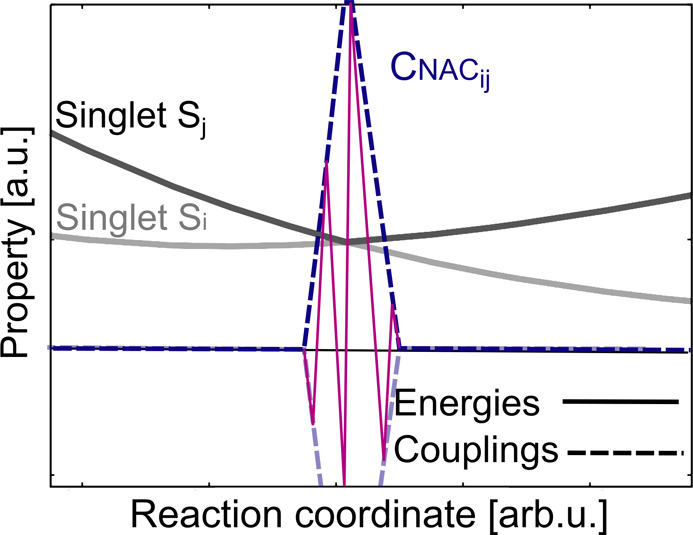
Electronically excited states of molecules are at the heart of photochemistry, photophysics, as well as photobiology and also play a role in material science. Their theoretical description requires highly accurate quantum chemical calculations, which are computationally expensive. In this review, we focus on not only how machine learning is employed to speed up such excited-state simulations but also how this branch of artificial intelligence can be used to advance this exciting research field in all its aspects. Discussed applications of machine learning for excited states include excited-state dynamics simulations, static calculations of absorption spectra, as well as many others. In order to put these studies into context, we discuss the promises and pitfalls of the involved machine learning techniques. Since the latter are mostly based on quantum chemistry calculations, we also provide a short introduction into excited-state electronic structure methods and approaches for nonadiabatic dynamics simulations and describe tricks and problems when using them in machine learning for excited states of molecules.
分子的电子激发态是光化学、光物理以及光生物学的核心,它们在材料科学中也起着重要作用。对其理论描述需要高度精确的量子化学计算,这在计算上是非常昂贵的。在这篇综述中,我们不仅关注机器学习如何被用来加速这种激发态模拟,还关注人工智能的这一分支如何在各个方面推进这个令人兴奋的研究领域。讨论的机器学习在激发态中的应用包括激发态动力学模拟、吸收光谱的静态计算以及许多其他方面。为了将这些研究置于上下文中,我们讨论了所涉及的机器学习技术的优点和缺点。由于后者主要基于量子化学计算,我们还提供了一个关于激发态电子结构方法和非绝热动力学模拟方法的简短介绍,并描述了在机器学习中使用它们来研究分子的激发态时的技巧和问题。