College of Medical and Dental Sciences, Institute of Cancer and Genomic Sciences, Centre for Computational Biology, University of Birmingham, Birmingham, B15 2TT, UK.
Institute of Translational Medicine, University Hospitals Birmingham NHS, Foundation Trust, Birmingham, B15 2TT, UK.
BMC Med Genomics. 2020 Nov 23;13(1):178. doi: 10.1186/s12920-020-00826-6.
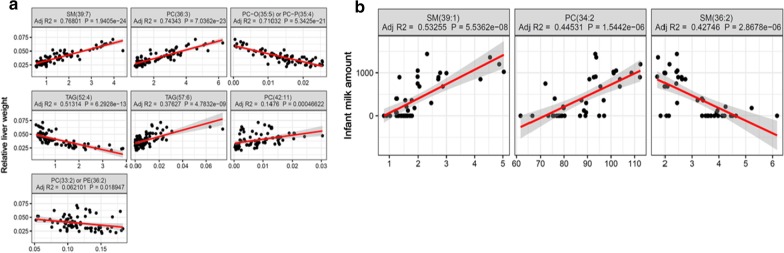
Biomarker identification is one of the major and important goal of functional genomics and translational medicine studies. Large scale -omics data are increasingly being accumulated and can provide vital means for the identification of biomarkers for the early diagnosis of complex disease and/or for advanced patient/diseases stratification. These tasks are clearly interlinked, and it is essential that an unbiased and stable methodology is applied in order to address them. Although, recently, many, primarily machine learning based, biomarker identification approaches have been developed, the exploration of potential associations between biomarker identification and the design of future experiments remains a challenge.
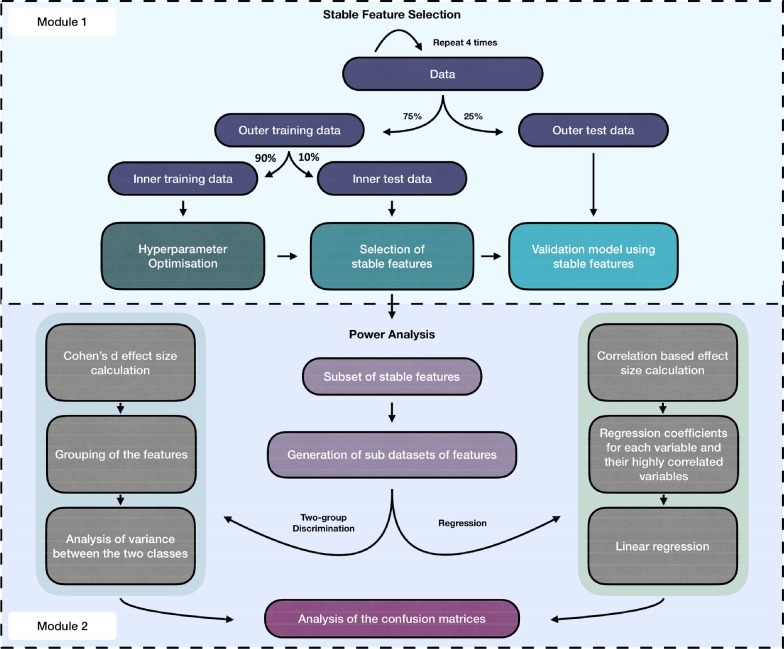
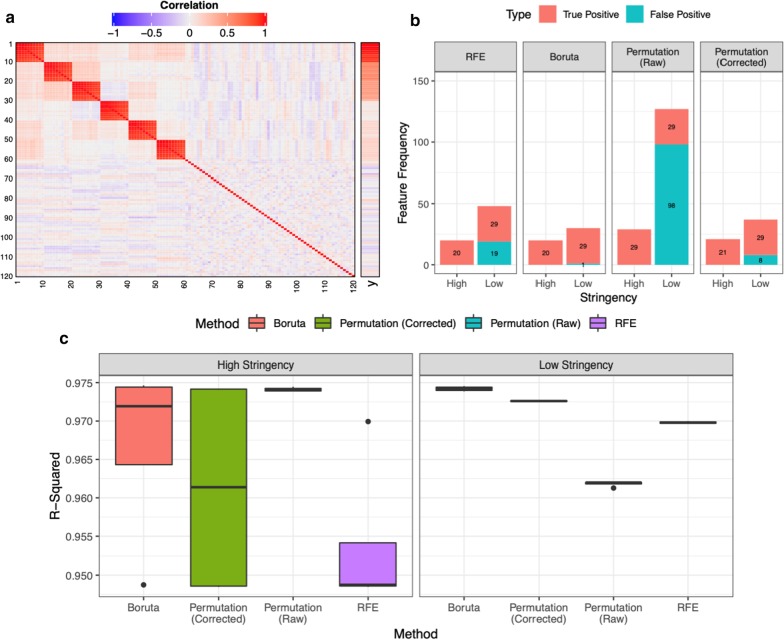
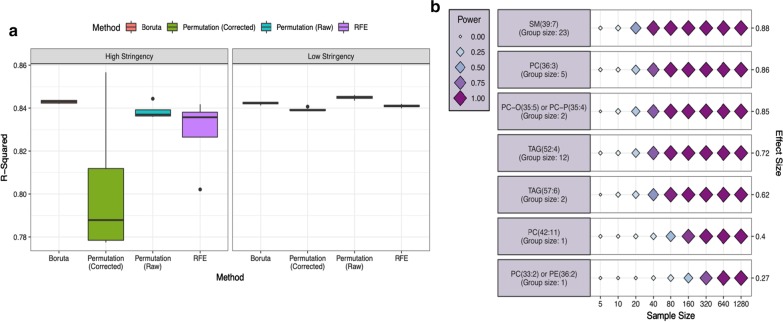
In this study, using both simulated and published experimentally derived datasets, we assessed the performance of several state-of-the-art Random Forest (RF) based decision approaches, namely the Boruta method, the permutation based feature selection without correction method, the permutation based feature selection with correction method, and the backward elimination based feature selection method. Moreover, we conducted a power analysis to estimate the number of samples required for potential future studies.
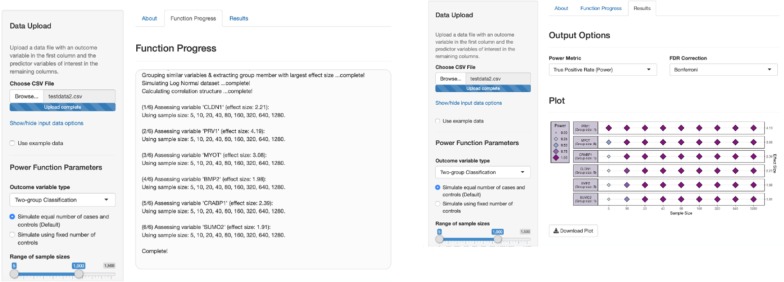
We present a number of different RF based stable feature selection methods and compare their performances using simulated, as well as published, experimentally derived, datasets. Across all of the scenarios considered, we found the Boruta method to be the most stable methodology, whilst the Permutation (Raw) approach offered the largest number of relevant features, when allowed to stabilise over a number of iterations. Finally, we developed and made available a web interface ( https://joelarkman.shinyapps.io/PowerTools/ ) to streamline power calculations thereby aiding the design of potential future studies within a translational medicine context.
We developed a RF-based biomarker discovery framework and provide a web interface for our framework, termed PowerTools, that caters the design of appropriate and cost-effective subsequent future omics study.
生物标志物的鉴定是功能基因组学和转化医学研究的主要和重要目标之一。大规模的组学数据不断积累,可以为复杂疾病的早期诊断和/或患者/疾病的高级分层提供生物标志物的鉴定提供重要手段。这些任务显然是相互关联的,为了解决这些问题,必须应用一种无偏且稳定的方法。尽管最近已经开发了许多主要基于机器学习的生物标志物鉴定方法,但探索生物标志物鉴定与未来实验设计之间的潜在关联仍然是一个挑战。
在这项研究中,我们使用模拟和已发表的实验衍生数据集,评估了几种最先进的基于随机森林 (RF) 的决策方法的性能,即 Boruta 方法、未校正的基于置换的特征选择方法、校正的基于置换的特征选择方法和基于后向消除的特征选择方法。此外,我们进行了功效分析,以估计潜在未来研究所需的样本数量。
我们提出了一些不同的基于 RF 的稳定特征选择方法,并使用模拟和已发表的实验衍生数据集比较了它们的性能。在所考虑的所有情况下,我们发现 Boruta 方法是最稳定的方法,而 Permutation(Raw)方法在允许经过多次迭代稳定后,提供了最多的相关特征。最后,我们开发并提供了一个网络界面(https://joelarkman.shinyapps.io/PowerTools/),以简化功效计算,从而有助于在转化医学背景下设计潜在的未来研究。
我们开发了一种基于 RF 的生物标志物发现框架,并提供了一个网络界面,称为 PowerTools,该界面可以为适当和具有成本效益的后续组学研究提供设计。