Tegally Houriiyah, Kensler Kevin H, Mungloo-Dilmohamud Zahra, Ghoorah Anisah W, Rebbeck Timothy R, Baichoo Shakuntala
Department of Digital Technologies, FoICDT, University of Mauritius, Réduit, Mauritius.
Dana Farber Cancer Institute, Harvard TH Chan School of Public Health, Boston, MA, United States of America.
PLoS One. 2020 Nov 24;15(11):e0242780. doi: 10.1371/journal.pone.0242780. eCollection 2020.
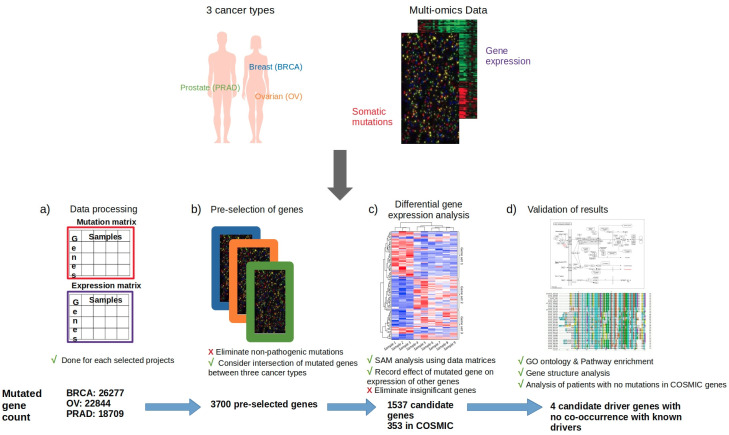
As the genomic profile across cancers varies from person to person, patient prognosis and treatment may differ based on the mutational signature of each tumour. Thus, it is critical to understand genomic drivers of cancer and identify potential mutational commonalities across tumors originating at diverse anatomical sites. Large-scale cancer genomics initiatives, such as TCGA, ICGC and GENIE have enabled the analysis of thousands of tumour genomes. Our goal was to identify new cancer-causing mutations that may be common across tumour sites using mutational and gene expression profiles. Genomic and transcriptomic data from breast, ovarian, and prostate cancers were aggregated and analysed using differential gene expression methods to identify the effect of specific mutations on the expression of multiple genes. Mutated genes associated with the most differentially expressed genes were considered to be novel candidates for driver mutations, and were validated through literature mining, pathway analysis and clinical data investigation. Our driver selection method successfully identified 116 probable novel cancer-causing genes, with 4 discovered in patients having no alterations in any known driver genes: MXRA5, OBSCN, RYR1, and TG. The candidate genes previously not officially classified as cancer-causing showed enrichment in cancer pathways and in cancer diseases. They also matched expectations pertaining to properties of cancer genes, for instance, showing larger gene and protein lengths, and having mutation patterns suggesting oncogenic or tumor suppressor properties. Our approach allows for the identification of novel putative driver genes that are common across cancer sites using an unbiased approach without any a priori knowledge on pathways or gene interactions and is therefore an agnostic approach to the identification of putative common driver genes acting at multiple cancer sites.
由于不同癌症患者的基因组特征因人而异,患者的预后和治疗可能因每个肿瘤的突变特征而有所不同。因此,了解癌症的基因组驱动因素并识别源自不同解剖部位的肿瘤之间潜在的突变共性至关重要。诸如TCGA、ICGC和GENIE等大规模癌症基因组计划使得对数千个肿瘤基因组进行分析成为可能。我们的目标是利用突变和基因表达谱识别可能在不同肿瘤部位普遍存在的新的致癌突变。汇总来自乳腺癌、卵巢癌和前列腺癌的基因组和转录组数据,并使用差异基因表达方法进行分析,以确定特定突变对多个基因表达的影响。与差异表达最显著的基因相关的突变基因被视为驱动突变的新候选基因,并通过文献挖掘、通路分析和临床数据调查进行验证。我们的驱动基因选择方法成功识别出了116个可能的新致癌基因,其中4个是在任何已知驱动基因均无改变的患者中发现的:MXRA5、OBSCN、RYR1和TG。先前未被正式归类为致癌的候选基因在癌症通路和癌症疾病中表现出富集。它们还符合对癌症基因特性的预期,例如,显示出更大的基因和蛋白质长度,并且具有表明致癌或抑癌特性的突变模式。我们的方法允许使用一种无偏倚的方法识别在不同癌症部位普遍存在的新的假定驱动基因,而无需对通路或基因相互作用有任何先验知识,因此是一种识别在多个癌症部位起作用的假定共同驱动基因的不可知方法。