University of Minnesota Genomics Center, Minneapolis, MN, 55455, USA.
Department of Genetics, Cell Biology, and Development, University of Minnesota, Minneapolis, MN, 55455, USA.
BMC Genomics. 2020 Dec 4;21(1):863. doi: 10.1186/s12864-020-07283-6.
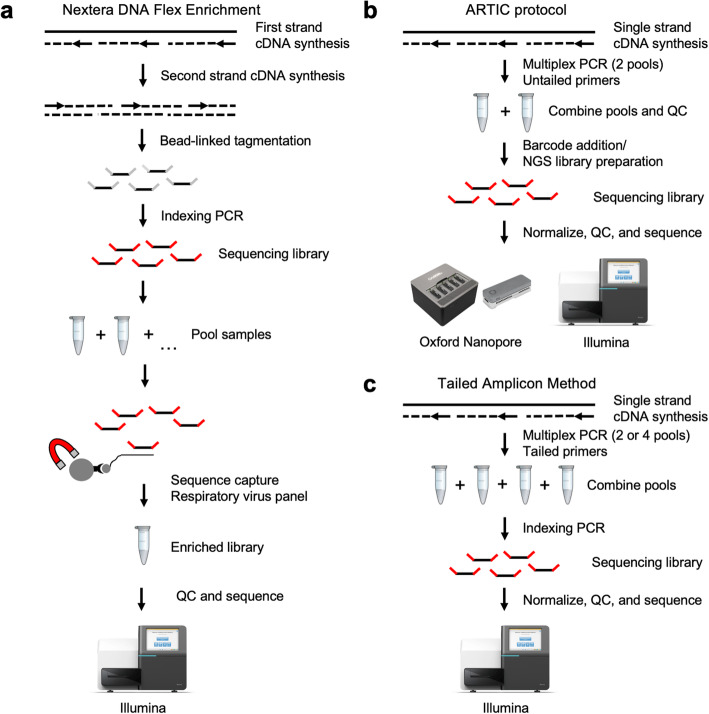
The global COVID-19 pandemic has led to an urgent need for scalable methods for clinical diagnostics and viral tracking. Next generation sequencing technologies have enabled large-scale genomic surveillance of SARS-CoV-2 as thousands of isolates are being sequenced around the world and deposited in public data repositories. A number of methods using both short- and long-read technologies are currently being applied for SARS-CoV-2 sequencing, including amplicon approaches, metagenomic methods, and sequence capture or enrichment methods. Given the small genome size, the ability to sequence SARS-CoV-2 at scale is limited by the cost and labor associated with making sequencing libraries.
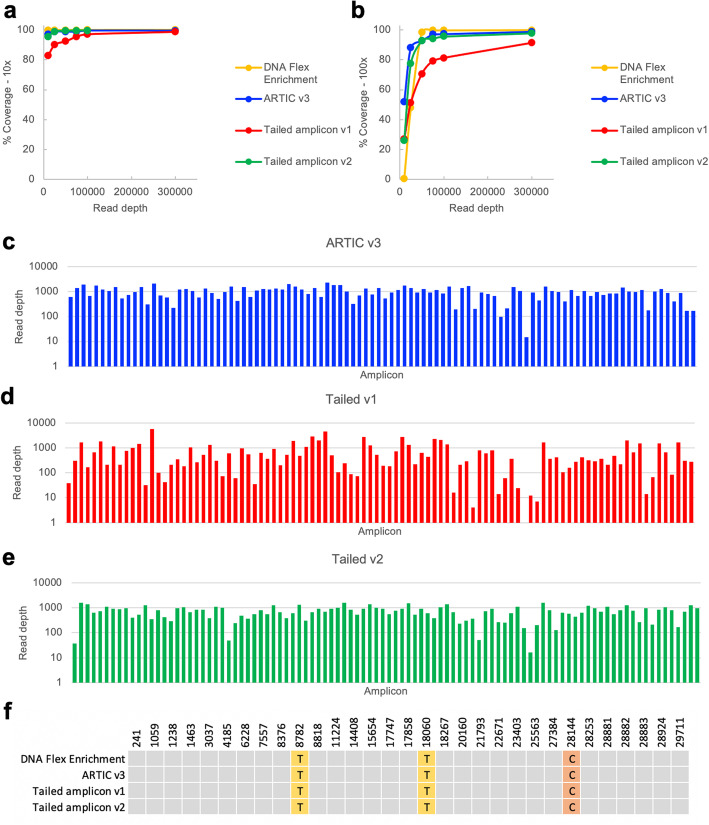
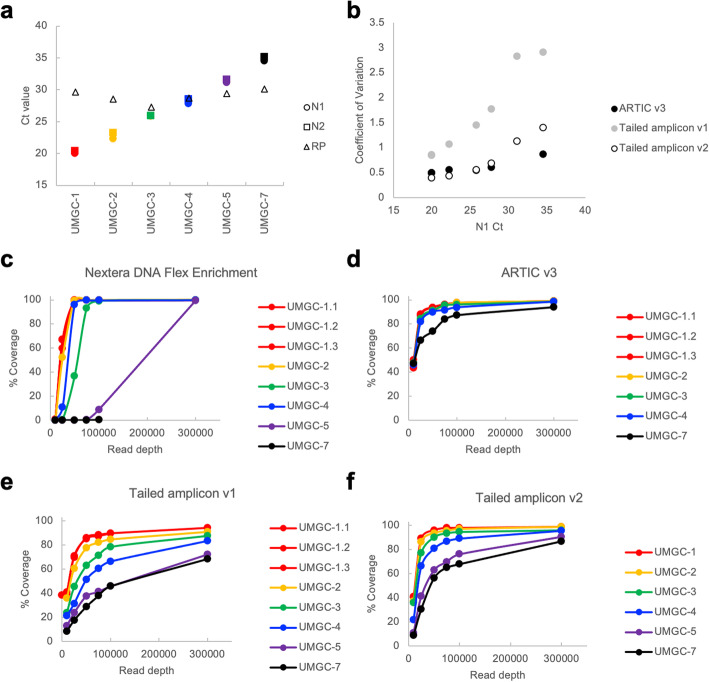
Here we describe a low-cost, streamlined, all amplicon-based method for sequencing SARS-CoV-2, which bypasses costly and time-consuming library preparation steps. We benchmark this tailed amplicon method against both the ARTIC amplicon protocol and sequence capture approaches and show that an optimized tailed amplicon approach achieves comparable amplicon balance, coverage metrics, and variant calls to the ARTIC v3 approach.
The tailed amplicon method we describe represents a cost-effective and highly scalable method for SARS-CoV-2 sequencing.
全球 COVID-19 大流行导致对临床诊断和病毒追踪的可扩展方法的迫切需求。下一代测序技术使对 SARS-CoV-2 的大规模基因组监测成为可能,因为全世界正在对数千个分离株进行测序,并将其存入公共数据存储库。目前正在使用短读长和长读长技术的多种方法来对 SARS-CoV-2 进行测序,包括扩增子方法、宏基因组方法以及序列捕获或富集方法。鉴于基因组较小,大规模测序 SARS-CoV-2 的能力受到与测序文库制备相关的成本和劳动力的限制。
在这里,我们描述了一种低成本、精简的、基于全扩增子的 SARS-CoV-2 测序方法,该方法绕过了昂贵且耗时的文库制备步骤。我们将这种长尾扩增子方法与 ARTIC 扩增子方案和序列捕获方法进行了基准测试,结果表明,优化后的长尾扩增子方法可实现与 ARTIC v3 方法相当的扩增子平衡、覆盖度指标和变异呼叫。
我们描述的长尾扩增子方法代表了一种具有成本效益且高度可扩展的 SARS-CoV-2 测序方法。