Department of Cell Biology, Harvard Medical School, Boston, Massachusetts, USA.
Department of Neuroscience, Genentech Inc, South San Francisco, California, USA.
J Biol Chem. 2021 Jan-Jun;296:100153. doi: 10.1074/jbc.RA120.015960. Epub 2020 Dec 10.
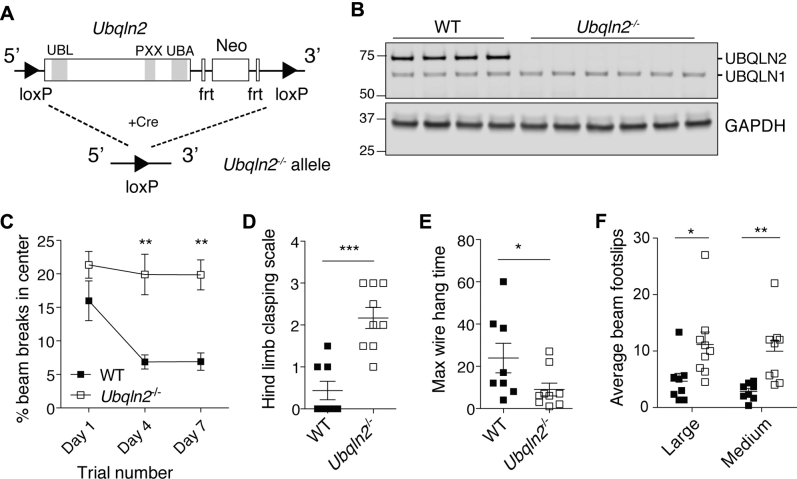
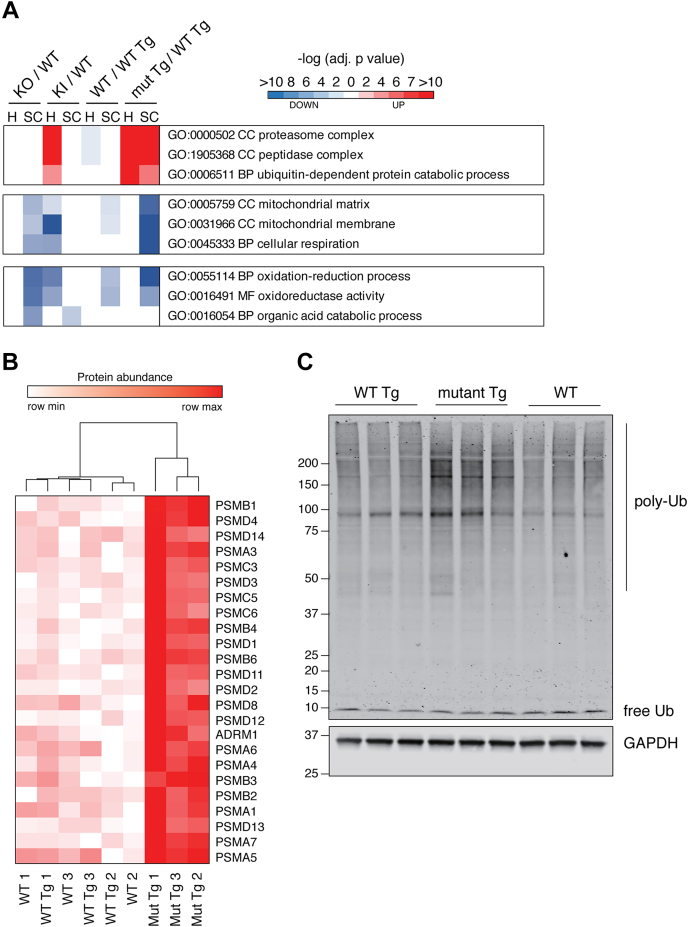
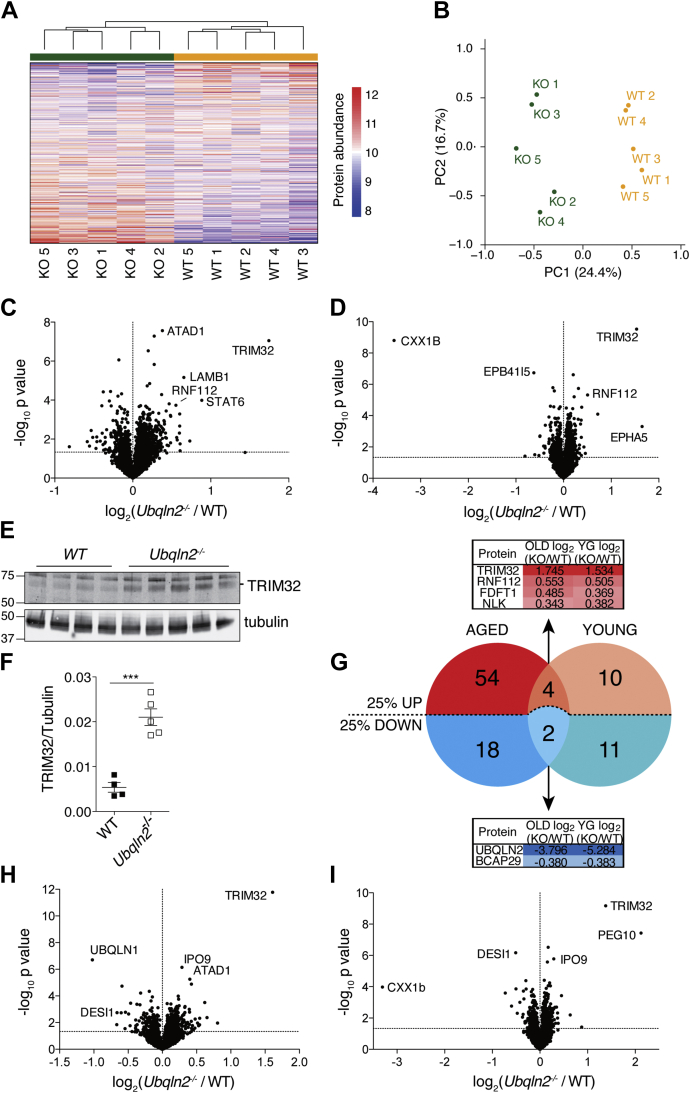
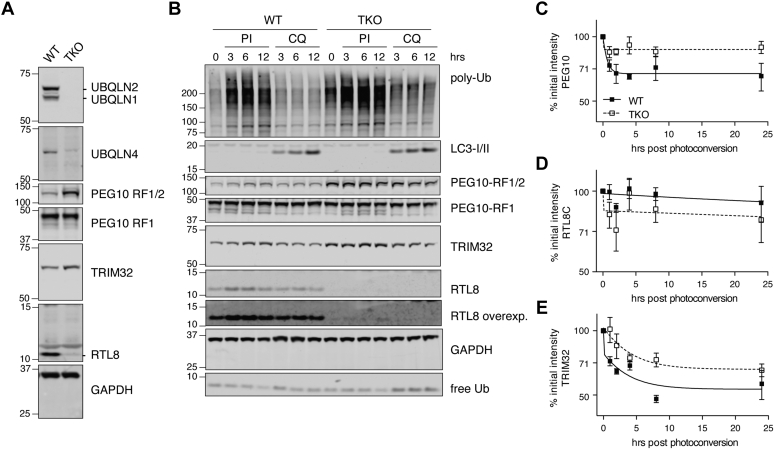
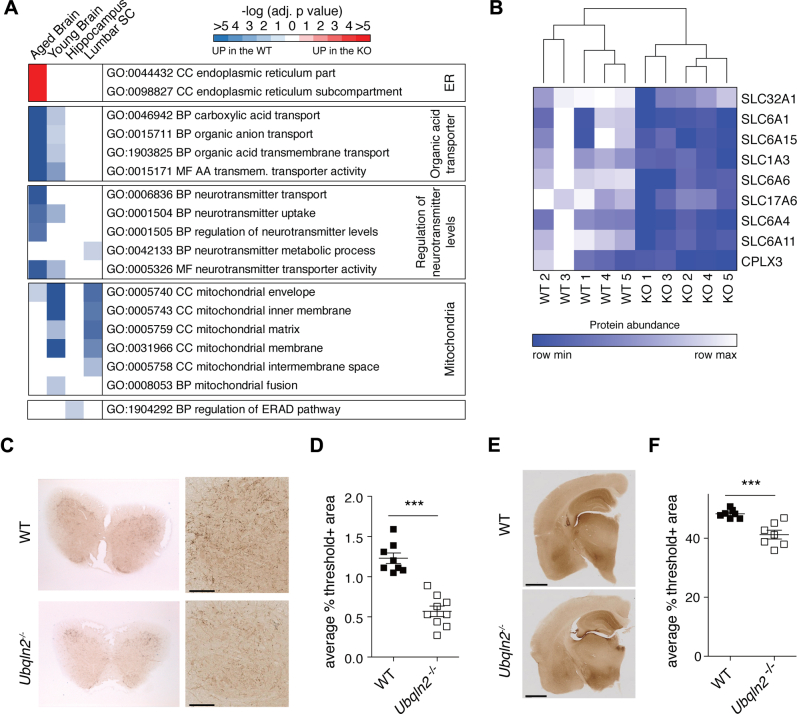
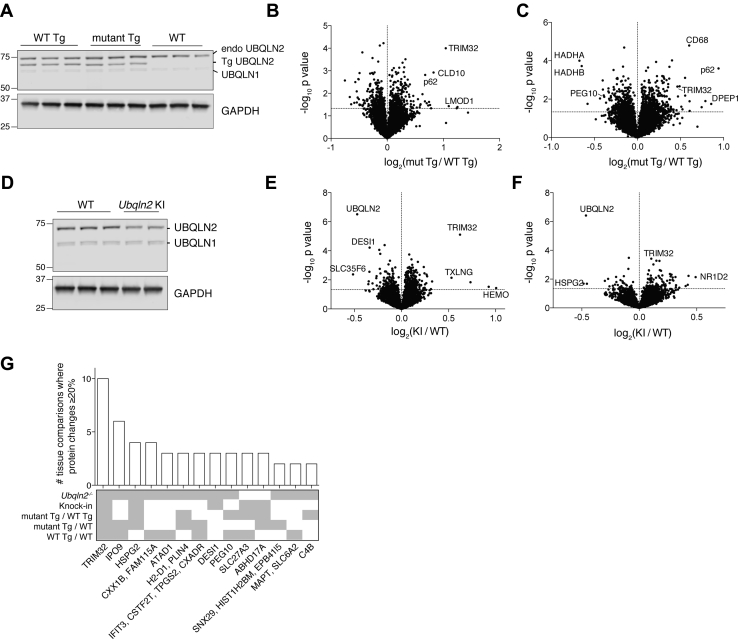
Familial neurodegenerative diseases commonly involve mutations that result in either aberrant proteins or dysfunctional components of the proteolytic machinery that act on aberrant proteins. UBQLN2 is a ubiquitin receptor of the UBL/UBA family that binds the proteasome through its ubiquitin-like domain and is thought to deliver ubiquitinated proteins to proteasomes for degradation. UBQLN2 mutations result in familial amyotrophic lateral sclerosis (ALS)/frontotemporal dementia in humans through an unknown mechanism. Quantitative multiplexed proteomics was used to provide for the first time an unbiased and global analysis of the role of Ubqln2 in controlling the composition of the proteome. We studied several murine models of Ubqln2-linked ALS and also generated Ubqln2 null mutant mice. We identified impacts of Ubqln2 on diverse physiological pathways, most notably serotonergic signaling. Interestingly, we observed an upregulation of proteasome subunits, suggesting a compensatory response to diminished proteasome output. Among the specific proteins whose abundance is linked to UBQLN2 function, the strongest hits were the ubiquitin ligase TRIM32 and two retroelement-derived proteins, PEG10 and CXX1B. Cycloheximide chase studies using induced human neurons and HEK293 cells suggested that PEG10 and TRIM32 are direct clients. Although UBQLN2 directs the degradation of multiple proteins via the proteasome, it surprisingly conferred strong protection from degradation on the Gag-like protein CXX1B, which is expressed from the same family of retroelement genes as PEG10. In summary, this study charts the proteomic landscape of ALS-related Ubqln2 mutants and identifies candidate client proteins that are altered in vivo in disease models and whose degradation is promoted by UBQLN2.
家族性神经退行性疾病通常涉及导致异常蛋白质或蛋白酶体功能成分异常的突变,这些功能成分作用于异常蛋白质。UBQLN2 是 UBL/UBA 家族的泛素受体,通过其泛素样结构域与蛋白酶体结合,被认为将泛素化蛋白递送至蛋白酶体进行降解。UBQLN2 突变通过未知机制导致人类家族性肌萎缩侧索硬化症(ALS)/额颞叶痴呆。定量多重蛋白质组学首次提供了对 Ubqln2 控制蛋白质组组成的作用的无偏和全面分析。我们研究了几种与 Ubqln2 相关的 ALS 小鼠模型,还生成了 Ubqln2 缺失突变体小鼠。我们确定了 Ubqln2 对多种生理途径的影响,尤其是 5-羟色胺能信号。有趣的是,我们观察到蛋白酶体亚基的上调,表明对蛋白酶体输出减少的代偿反应。在与 UBQLN2 功能相关的特定蛋白质中,丰度最高的是泛素连接酶 TRIM32 和两个逆转录元件衍生蛋白 PEG10 和 CXX1B。使用诱导的人神经元和 HEK293 细胞进行的环己酰亚胺追踪研究表明,PEG10 和 TRIM32 是直接的客户。尽管 UBQLN2 通过蛋白酶体指导多种蛋白质的降解,但它令人惊讶地赋予 Gag 样蛋白 CXX1B 很强的保护作用,使其免受降解,CXX1B 是从与 PEG10 相同的逆转录元件基因家族表达的。总之,这项研究绘制了与 ALS 相关的 Ubqln2 突变体的蛋白质组图谱,并确定了候选客户蛋白,这些蛋白在疾病模型中体内发生改变,其降解受 UBQLN2 促进。